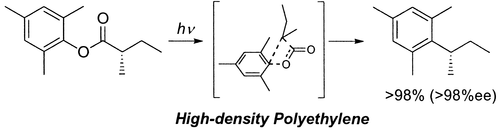
Photodecarboxylation is the exclusive photoreaction of 2,4,6-trimethylphenyl (S)-2-methylbutanoate in unstretched high-density polyethylene films. The sole product, (S)-1-(2-methylpropyl)-2,4,6-trimethylbenzene, is formed with complete retention of stereochemistry. In other polyethylene films, organic solvents, and β-cyclodextrin cavities, cage-escaped products derived from Fries-type bond scission are obtained as well. The results indicate the importance of the media in controlling the conformations of aryl esters and, thereby, their photoreactions.
@article{mori2003enhanced,title={Enhanced photodecarboxylation of an aryl ester in polyethylene films},author={Mori, Tadashi and Inoue, Yoshihisa and Weiss, Richard G},journal={Org. Lett.},volume={5},number={24},pages={4661--4664},year={2003},publisher={ACS Publications},doi={10.1021/ol035852t},url={https://doi.org/10.1021/ol035852t},dimensions={true},tab={paper},}
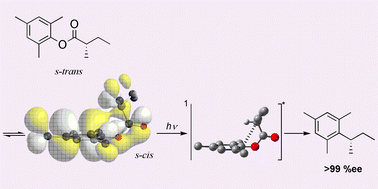
Complete memory of chirality upon photodecarboxylation of mesityl alkanoate to mesitylalkane: theoretical and experimental evidence for cheletropic decarboxylation via a spiro-lactonic transition state
Tadashi Mori*, Hideaki Saito, and Yoshihisa Inoue*
The photodecarboxylation of chiral mesityl alkanoate to mesitylalkane has been studied experimentally/theoretically, and it has been found that the photodecarboxylation proceeds to give the product in >99% enantiomeric excesses under a variety of conditions, indicating no involvement of any radical intermediates, but that the reaction proceeds through the concerted cheletropic extrusion of CO2 from the energetically less-favored s-cis conformation.
@article{mori2003complete,title={Complete memory of chirality upon photodecarboxylation of mesityl alkanoate to mesitylalkane: theoretical and experimental evidence for cheletropic decarboxylation via a spiro-lactonic transition state},author={Mori, Tadashi and Saito, Hideaki and Inoue, Yoshihisa},journal={Chem. Commun.},number={18},pages={2302--2303},year={2003},publisher={Royal Society of Chemistry},doi={10.1039/b305267b},url={https://doi.org/10.1039/b305267b},dimensions={true},tab={paper},}
Bovine serum albumin-mediated enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate
Takehiko Wada, Masaki Nishijima, Tai Fujisawa, Norimitsu Sugahara, Tadashi Mori, Asao Nakamura, and Yoshihisa Inoue*
Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxyalate (AC) was performed at 25 °C in aqueous buffer solution (pH 7) in the presence of bovine-serum albumin (BSA) to afford four [4 + 4] cyclodimers, i.e., anti- and syn-head-to-tail (HT) (1 and 2) and anti- and syn-head-to-head (HH) dimers (3 and 4), of which only 2 and 3 are chiral. We found that (1) BSA possesses four sets of binding sites for AC of different affinities, stoichiometries, and chiral environment for photoreaction, which bind 1, 3, 2, and 3 AC molecules with binding constants of 5.3 × 107, 1.3 × 105, 1.4 × 104, and 3.0 × 103 M-1, respectively, (2) the regioselectivity of photodimerization is switched from HT to HH by adding BSA (the HH/HT ratio varies from 0.28 to 4.3), (3) BSA-mediated photodimerization of AC affords optically active products 2 and 3 of up to 29% and 41% ee, respectively. It is emphasized that the selective excitation of bound substrate, utilizing the spectral shift upon complexation with BSA, is not a prerequisite for efficient photochirogenesis using biomolecules.
@article{wada2003bovine,title={Bovine serum albumin-mediated enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate},author={Wada, Takehiko and Nishijima, Masaki and Fujisawa, Tai and Sugahara, Norimitsu and Mori, Tadashi and Nakamura, Asao and Inoue, Yoshihisa},journal={J. Am. Chem. Soc.},volume={125},number={25},pages={7492--7493},year={2003},publisher={ACS Publications},doi={10.1021/ja034641g},url={https://doi.org/10.1021/ja034641g},dimensions={true},tab={paper},}
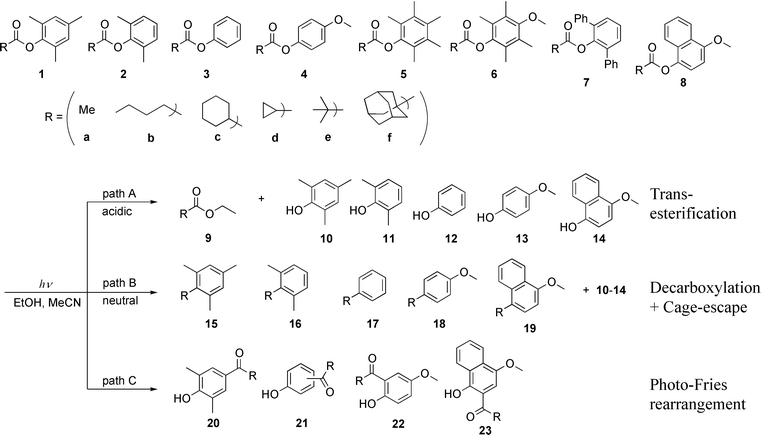
Acid-controlled photoreactions of aryl alkanoates: competition of transesterification, decarboxylation, Fries-rearrangement and/or transposition
Tadashi Mori*, Makoto Takamoto, Takehiko Wada, and Yoshihisa Inoue*
2,4,6-Trimethylphenyl (mesityl) cyclohexanecarboxylate 1c and related mesityl esters 1a,b and d–f were photodecarboxylated upon irradiation at 254 nm in neutral solvents to give alkylmesitylenes 15 in good yields. In contrast, in the presence of a catalytic amount of acid and a sufficient amount of alcohol, the same compounds underwent facile phototransesterification to afford the corresponding ester 9 and phenol 10 in almost quantitative yields. Photolyses of less-substituted aryl alkanoates such as 2,6-dimethylphenyl (xylenyl), phenyl, 4-methoxyphenyl (4-anisyl) and 4-methoxynaphthyl alkanoates 2–4 and 8 were also investigated to elucidate the reaction mechanism of acid-catalyzed phototransesterification. Irradiation of these esters afforded the corresponding photo-Fries rearrangement products 20–23 both in neutral and acidic acetonitrile with significant acceleration of the processes in the presence of acid. It was elucidated that the photolysis of the esters affords the geminate radical pair 28, which in turn recombines to cyclohexadienone intermediates 29 and 30. Added acid accelerates not only the nucleophilic attack of alcohol to 29 and 30 but also other processes. Prolonged irradiation of the esters in neutral solution led to skeletal rearrangements of the initial product, affording isomeric alkylbenzenes. The phototransposition of cyclohexylmesitylene 15cvia benzvalene intermediates was, on the contrary, retarded under acidic conditions. These acid-controlled competitive photoreactions are representative examples, in which a catalytic amount of acid can alter the fate of reactive intermediates on both ground-state and excited-state surfaces.
@article{mori2003acid,title={Acid-controlled photoreactions of aryl alkanoates: competition of transesterification, decarboxylation, Fries-rearrangement and/or transposition},author={Mori, Tadashi and Takamoto, Makoto and Wada, Takehiko and Inoue, Yoshihisa},journal={Photochem. Photobiol. Sci.},volume={2},number={11},pages={1187--1199},year={2003},publisher={Royal Society of Chemistry},doi={10.1039/b305898k},url={https://doi.org/10.1039/b305898k},dimensions={true},tab={paper},}
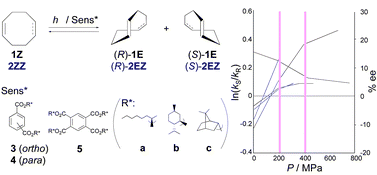
Pressure control of enantiodifferentiating photoisomerization of cyclooctenes sensitized by chiral benzenepolycarboxylates. The origin of discontinuous pressure dependence of the optical yield
Pressure effects on enantiodifferentiating geometrical photoisomerizations of (Z)-cyclooctene and (Z,Z)-cycloocta-1,5-diene sensitized by chiral benzene-1,2,4,5-tetracarboxylate were investigated over a pressure range of 0.1–750 MPa. Enantiomeric excesses (ee’s) of the (E)- and (E,Z)-isomers obtained displayed discontinuous pressure dependencies, affording distinctly different differential activation volumes (ΔΔV‡) for each range, indicating alteration of the enantiodifferentiation mechanism. The switching of ΔΔV‡ occurred at essentially the same pressures of 200 and 400 MPa, which are shared by all the chiral sensitizers, irrespective of the chiral auxiliary employed. Circular dichroism spectral examinations at pressures of up to 400 MPa also revealed that the chiral sensitizers undergo discontinuous conformational changes at 200 MPa, which most likely lead to switching of the enantiodifferentiating sensitization mechanism in the exciplex intermediate.
@article{kaneda2003pressure,title={Pressure control of enantiodifferentiating photoisomerization of cyclooctenes sensitized by chiral benzenepolycarboxylates. The origin of discontinuous pressure dependence of the optical yield},author={Kaneda, Masayuki and Nakamura, Asao and Asaoka, Sadayuki and Ikeda, Haruhiko and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa},journal={Org. Biomol. Chem.},volume={1},number={24},pages={4435--4440},year={2003},publisher={Royal Society of Chemistry},doi={10.1039/b310491e},url={https://doi.org/10.1039/b310491e},dimensions={true},tab={paper},}