articles
公表論文
original papers / accounts & reviews / cover pictures
2026 / 2025 / 2024 / 2023 / 2022 / 2021 / 2020 / 2019 / 2018 / 2017 / 2016 / 2015 / 2014 / 2013 / 2012 / 2011 / 2010 / 2009 / 2008 / 2007 / 2006 / 2005 / 2004 / 2003 / 2002 / 2001 / 2000 / 1999 / 1998 / 1997 / 1996 / 1995
2026
-
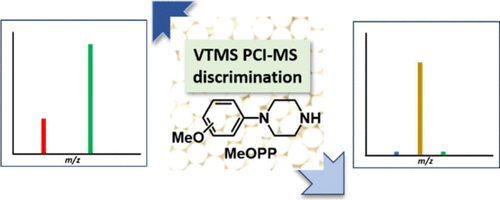 Vinyltrimethylsilane Chemical Ionization Tandem Mass Spectrometry Separates Methoxyphenylpiperazine IsomersZiaho Qin, Shinji Tsunoi*, Koya Hachiri, Ikuya Shibata, and Tadashi Mori*Anal. Chem., 2026, in press.
Vinyltrimethylsilane Chemical Ionization Tandem Mass Spectrometry Separates Methoxyphenylpiperazine IsomersZiaho Qin, Shinji Tsunoi*, Koya Hachiri, Ikuya Shibata, and Tadashi Mori*Anal. Chem., 2026, in press.Novel synthetic drugs often contain aromatic positional isomers that are difficult to distinguish by conventional mass spectrometry due to similar fragmentation patterns and retention times. Isomers of methoxyphenylpiperazine (MeOPPs) exemplify this analytical challenge. Here, we employed chemical ionization tandem mass spectrometry using vinyltrimethylsilane as the reagent gas to analyze trifluoroacetyl-derivatized MeOPP isomers. This approach generated diagnostic fragment ions unique to the ortho, meta, and para isomers, enabling their unambiguous discrimination in a single analytical run. Density functional theory calculations supported the proposed fragmentation pathways and provided mechanistic insight into the observed isomer-specific selectivity. This straightforward and robust method offers a promising strategy for precise isomer identification in forensic and pharmaceutical contexts where the accurate differentiation of synthetic drug isomers is critical.
@article{qin2026vinyltrimethylsilane, title = {Vinyltrimethylsilane Chemical Ionization Tandem Mass Spectrometry Separates Methoxyphenylpiperazine Isomers}, author = {Qin, Ziaho and Tsunoi, Shinji and Hachiri, Koya and Shibata, Ikuya and Mori, Tadashi}, journal = {Anal. Chem.}, pages = {in press}, year = {2026}, month = mar, publisher = {ACS Publications}, doi = {10.1021/acs.analchem.5c08125}, url = {https://doi.org/10.1021/acs.analchem.5c08125}, dimensions = {true}, tab = {paper}, } -
 Small molecule helical emittersTadashi Mori*Chem. Soc. Rev., 2026, 55, 1999–2023.
Small molecule helical emittersTadashi Mori*Chem. Soc. Rev., 2026, 55, 1999–2023.The development of materials exhibiting circularly polarized luminescence (CPL) is a key area of research for next-generation optical technologies, including 3D displays and secure communications. The central goal in this field is to create chiral emitters with a high luminescence dissymmetry (gCPL) factor, a measure of the emission’s chirality. While theoretically reaching ±2, practical values in small organic molecules have historically been much lower, on the order of 0.001 or less. This summary outlines the core strategies in molecular design focusing on helical emitters that have recently enabled significant breakthroughs, pushing g values beyond the 0.01 threshold. The magnitude of g factor is determined by the cosine of the angle between the electric (μe) and magnetic (μm) dipole transition moments, as well as their respective magnitudes. Consequently, the most successful research has moved beyond simple screening and has focused on rationally engineering molecules to optimize this relationship. One of the most direct strategies has been to design rigid, helical molecules where high symmetry forces the μe and μm to be parallel. By enforcing D2 and other symmetry in certain helicenes, helical nanographenes and related structures, researchers have minimized the angle between the moments, thus maximizing the cosine term and leading to a significant enhancement in the g factor value. A second, distinct approach targets the magnitude of the μm. In most organic chromophores, μm is inherently small, limiting the potential g factor intensity. To overcome this, researchers have designed for example belt-shaped macrocyclic molecules that function as molecular-scale solenoids. The cyclic arrangement of chromophores induces a large, circulating electric current in the excited state, which in turn generates a powerful μm along the cylinder’s axis. A third innovative strategy circumvents the limitation of a small intrinsic μm by leveraging exciton coupling between two and more chromophores. In these systems, two π-conjugated units such as pyrene are held in a fixed, chiral arrangement. Upon photoexcitation, they form an intramolecular excimer, a transient excited-state complex with a well-defined helical geometry. The resulting CPL signal originates from the chiral interaction of the two strong electric transition moments, generating a large rotational strength and a high g factor without relying on the weak magnetic moment of the individual units. The progress in CPL-active materials is a testament to the power of targeted molecular engineering. As seen in the state-of-the-art examples in the review, the field has matured to a point where the fundamental photophysical principles governing CPL are being directly translated into synthetic molecular designs. While current high-performing materials are often complex and synthetically challenging, these proof-of-concept molecules validate the core design strategies.
@article{mori2026small, title = {Small molecule helical emitters}, author = {Mori, Tadashi}, journal = {Chem. Soc. Rev.}, pages = {1999--2023}, year = {2026}, volume = {55}, issue = {4}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d5cs01270h}, url = {https://doi.org/10.1039/d5cs01270h}, dimensions = {true}, tab = {review}, }
2025
-
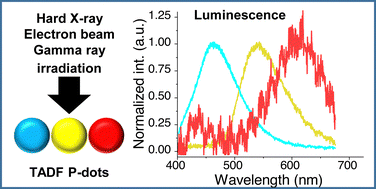 Color variation in radio-luminescence of P-dots doped with thermally activated delayed fluorescence moleculesZheming Su, Hieu Thi Minh Nguyen, Zuoyue Liu, Daiki Asanuma, Minoru Yamaji, Masanori Koshimizu, Hajime Shigemitsu, Sachiko Tojo, Tadashi Mori, Toshiyuki Kida, Guillem Pratx*, Mamoru Fujitsuka*, and Yasuko Osakada*Phys. Chem. Chem. Phys., 2025, 27, 7605–7610.
Color variation in radio-luminescence of P-dots doped with thermally activated delayed fluorescence moleculesZheming Su, Hieu Thi Minh Nguyen, Zuoyue Liu, Daiki Asanuma, Minoru Yamaji, Masanori Koshimizu, Hajime Shigemitsu, Sachiko Tojo, Tadashi Mori, Toshiyuki Kida, Guillem Pratx*, Mamoru Fujitsuka*, and Yasuko Osakada*Phys. Chem. Chem. Phys., 2025, 27, 7605–7610.Thermally activated delayed fluorescence (TADF) materials possess exceptional photophysical properties. Organic scintillators utilizing TADF materials have shown great promise for applications requiring efficient radio-luminescence, owing to their high quantum efficiency and tunable emission properties. Previous studies demonstrated that polymer dots (P-dots) doped with TADF materials exhibit radio-luminescence under hard X-ray and electron beam excitation. However, the TADF materials used in these experiments were limited to limited color options, restricting their utility and hindering the exploration of multicolor radio-luminescence necessary for advanced applications. In this study, we successfully achieved multicolor radio-luminescence-blue, yellow, and red-by developing P-dots doped with TADF materials that emit across the visible spectrum. This breakthrough was demonstrated under excitation by hard X-rays, gamma rays, and electron beams. The ability to realize multicolor radio-luminescence is crucial, as it enables enhanced spatial and spectral resolution, which is vital for applications such as high-precision bio-imaging and multimodal sensing.
@article{su2025color, title = {Color variation in radio-luminescence of P-dots doped with thermally activated delayed fluorescence molecules}, author = {Su, Zheming and Nguyen, Hieu Thi Minh and Liu, Zuoyue and Asanuma, Daiki and Yamaji, Minoru and Koshimizu, Masanori and Shigemitsu, Hajime and Tojo, Sachiko and Mori, Tadashi and Kida, Toshiyuki and Pratx, Guillem and Fujitsuka, Mamoru and Osakada, Yasuko}, journal = {Phys. Chem. Chem. Phys.}, volume = {27}, number = {15}, pages = {7605--7610}, year = {2025}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d5cp00410a}, url = {https://doi.org/10.1039/d5cp00410a}, dimensions = {true}, tab = {paper}, } -
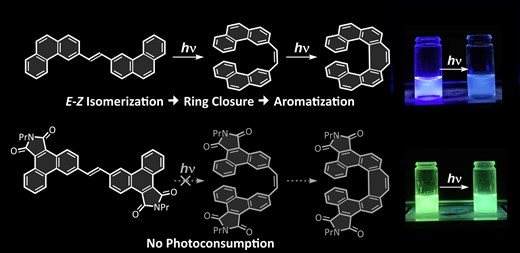 Highly emissive and robust diarylethene fluorophore incorporating imide-fused phenanthryl moietiesKeito Nose, Minoru Yamaji, Tadashi Mori, Fumito Tani, Kenta Goto, and Hideki Okamoto*Chem. Lett., 2025, 54, upaf091.
Highly emissive and robust diarylethene fluorophore incorporating imide-fused phenanthryl moietiesKeito Nose, Minoru Yamaji, Tadashi Mori, Fumito Tani, Kenta Goto, and Hideki Okamoto*Chem. Lett., 2025, 54, upaf091.The fluorescence properties and photostabilities of (E)-diarylethenes (DPEs), featuring two 3-phenanthryl moieties, were investigated. Pristine (E)-di-1,2-(3-phenanthryl)ethene and its ester derivative, respectively referred to as DPE-H and DPE-E, exhibited blue fluorescence. However, they underwent a two-step photoreaction sequence that yielded [7]helicenes, resulting in a gradual reduction of the fluorescence intensity. In contrast, the imide-fused derivative DPE-I displayed intense green fluorescence and it was unexpectedly photostable in solution, maintaining its highly efficient fluorescent nature even after prolonged light exposure. DPE-I, thus, provides a new type of highly efficient and robust diarylethene fluorophore.
@article{nose2025highly, title = {Highly emissive and robust diarylethene fluorophore incorporating imide-fused phenanthryl moieties}, author = {Nose, Keito and Yamaji, Minoru and Mori, Tadashi and Tani, Fumito and Goto, Kenta and Okamoto, Hideki}, journal = {Chem. Lett.}, volume = {54}, number = {5}, pages = {upaf091}, year = {2025}, publisher = {Oxford University Press UK}, doi = {10.1093/chemle/upaf091}, url = {https://doi.org/10.1093/chemle/upaf091}, dimensions = {true}, tab = {paper}, } -
 Inversion of circularly polarized luminescence by electric current flow during transitionAyumi Imayoshi*, Shinya Fujio, Yuuki Nagaya, Misato Sakai, Atsushi Terazawa, Misa Sakura, Keita Okada, Takahiro Kimoto, Tadashi Mori, Yoshitane Imai, Masahiko Hada, and Kazunori Tsubaki*Phys. Chem. Chem. Phys., 2025, 27, 77–82.
Inversion of circularly polarized luminescence by electric current flow during transitionAyumi Imayoshi*, Shinya Fujio, Yuuki Nagaya, Misato Sakai, Atsushi Terazawa, Misa Sakura, Keita Okada, Takahiro Kimoto, Tadashi Mori, Yoshitane Imai, Masahiko Hada, and Kazunori Tsubaki*Phys. Chem. Chem. Phys., 2025, 27, 77–82.The development of chiral compounds exhibiting circularly polarized luminescence (CPL) has advanced remarkably in recent years. Designing CPL-active compounds requires an understanding of the electric transition dipole moment (μ) and the magnetic transition dipole moment (m) in the excited state. However, while the direction and magnitude of μ can, to some extent, be visually inferred from chemical structures, m remains elusive, posing challenges for direct predictions based on structural information. This study utilized binaphthol, a prominent chiral scaffold, and achieved CPL-sign inversion by strategically varying the substitution positions of phenylethynyl (PE) groups on the binaphthyl backbone, while maintaining consistent axial chirality. Theoretical investigation revealed that the substitution position of PE groups significantly affects the orientation of m in the excited state, leading to CPL-sign inversion. Furthermore, we propose that this CPL-sign inversion results from a reversal in the rotation of instantaneous current flow during the S1 → S0 transition, which in turn alters the orientation of m. The current flow can be predicted from the chemical structure, allowing anticipation of the properties of m and, consequently, the characteristics of CPL. This insight provides a new perspective in designing CPL-active compounds, particularly for C2-symmetric molecules where the S1 -> S0 transition predominantly involves LUMO → HOMO transitions. If μ represents the directionality of electron movement during transitions, i.e., the “difference” in electron locations before and after transitions, then m could be represented as the “path” of electron movement based on the current flow during the transition.
@article{imayoshi2025inversion, title = {Inversion of circularly polarized luminescence by electric current flow during transition}, author = {Imayoshi, Ayumi and Fujio, Shinya and Nagaya, Yuuki and Sakai, Misato and Terazawa, Atsushi and Sakura, Misa and Okada, Keita and Kimoto, Takahiro and Mori, Tadashi and Imai, Yoshitane and Hada, Masahiko and Tsubaki, Kazunori}, journal = {Phys. Chem. Chem. Phys.}, volume = {27}, number = {1}, pages = {77--82}, year = {2025}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d4cp02968b}, url = {https://doi.org/10.1039/d4cp02968b}, dimensions = {true}, tab = {paper}, }
2024
-
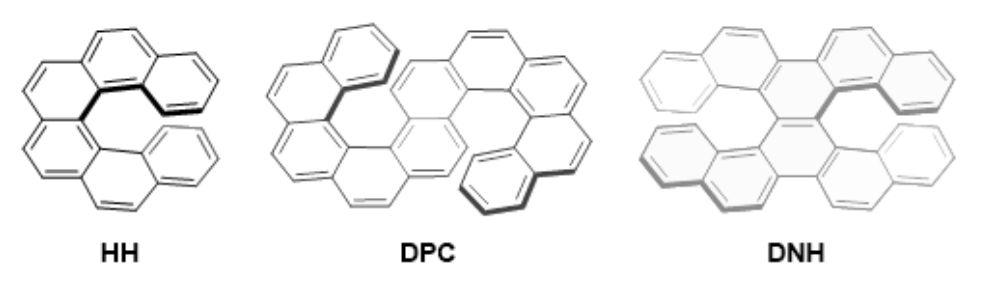
 Significance of Vibronic Coupling that Shapes Circularly Polarized Luminescence of Double HelicenesTadashi Mori*Angew. Chem. Int. Ed., 2024, 63, e202402960.
Significance of Vibronic Coupling that Shapes Circularly Polarized Luminescence of Double HelicenesTadashi Mori*Angew. Chem. Int. Ed., 2024, 63, e202402960.The circularly polarized luminescence (CPL) spectra of S- and X-shaped double helicenes exhibit distinct vibrational structures and overall shape variations. In this study, we conducted an in-depth investigation into the vibronic effects influencing the CPL spectra of two double helicenes, namely DPC and DNH. Employing state-of-the-art computations utilizing the FC-HT1|VH model at the CAM−B3LYP/def2-TZVP level, we unveiled the paramount impact of Franck–Condon (FC), Herzberg-Teller (HT), and Duschinsky effects on their chiroptical responses. Our research underscores the pivotal role of structural deformations associated with the S1-to-S0 electronic transition in molding CPL spectra and wavelength-dependent dissymmetry (g) factor values, as well as the significance of HT effects in shaping and enhancing CPL responses. This extensive investigation not only advances our comprehension of the vibronic characteristics in configurationally distinct double helicenes but also offers valuable insights for the design of chiral molecules featuring controllable or finely-tunable CPL responses.
@article{mori2024significance, title = {Significance of Vibronic Coupling that Shapes Circularly Polarized Luminescence of Double Helicenes}, author = {Mori, Tadashi}, journal = {Angew. Chem. Int. Ed.}, volume = {63}, number = {16}, pages = {e202402960}, year = {2024}, publisher = {Wiley Online Library}, doi = {10.1002/anie.202319702}, url = {https://doi.org/10.1002/anie.202319702}, dimensions = {true}, tab = {paper}, } -
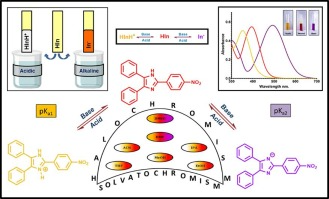 Solvatochromism and halochromism in nitro lophine derivatives: Photophysical study, computational calculations and applications as pH sensing materialShiv R Desai, Rinkal B Bhoraniya, Mahesh Koladiya, Vidhi V Bhopekar, Chintan R Patel, Tadashi Mori, and Sachin G Modha*J. Photochem. Photobiol. A: Chem., 2024, 455, 115751.
Solvatochromism and halochromism in nitro lophine derivatives: Photophysical study, computational calculations and applications as pH sensing materialShiv R Desai, Rinkal B Bhoraniya, Mahesh Koladiya, Vidhi V Bhopekar, Chintan R Patel, Tadashi Mori, and Sachin G Modha*J. Photochem. Photobiol. A: Chem., 2024, 455, 115751.Nitro Lophines were readily synthesized via multicomponent reaction of benzil, nitro benzaldehyde and ammonium acetate. All three nitro Lophines were isolated in good yields and were also characterized by various analytical and spectroscopic techniques. A distinctive solvatochromism was observed in case of ortho and para nitro containing Lophines in their absorbance spectra. In particular, naked eye solvatochromism was observed, in case of para nitro Lophine, in the form of yellow and red coloured solution in methanol and DMSO respectively. Halochromism studies revealed the hypsochromic shift under acidic pH while bathochromic shift under alkaline pH for both nitro Lophines. Again the para nitro Lophine demonstrated wonderful distinctive colour change under acidic and alkaline pH in both methanol and DMSO as solvents. The measurement of absorbance at different pH values was carried out and the data obtained were used for calculation of dissociation constants employing several graphical methods. Computational calculations were carried out using TD-DFT for UV–Vis absorption calculation while graph-convolutional neural network model was utilized for calculation of dissociation constants. Ultimately, a reusable naked eye pH detection paper could be developed using para nitro Lophine as a sole indicator.
@article{desai2024solvatochromism, title = {Solvatochromism and halochromism in nitro lophine derivatives: Photophysical study, computational calculations and applications as pH sensing material}, author = {Desai, Shiv R and Bhoraniya, Rinkal B and Koladiya, Mahesh and Bhopekar, Vidhi V and Patel, Chintan R and Mori, Tadashi and Modha, Sachin G}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {455}, pages = {115751}, year = {2024}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2024.115751}, url = {https://doi.org/10.1016/j.jphotochem.2024.115751}, dimensions = {true}, tab = {paper}, } -
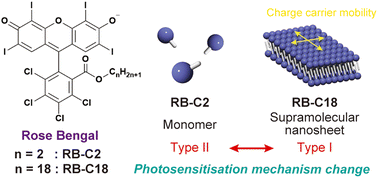 Supramolecular nanosheet formation-induced photosensitisation mechanism change of Rose Bengal dye in aqueous mediaAsuka Bunno, Hajime Shigemitsu*, Aya Yoshikawa, Yasuko Osakada, Mamoru Fujitsuka, Fumitaka Ishiwari, Akinori Saeki, Kei Ohkubo, Tadashi Mori, and Toshiyuki Kida*Chem. Commun., 2024, 60, 889–892.
Supramolecular nanosheet formation-induced photosensitisation mechanism change of Rose Bengal dye in aqueous mediaAsuka Bunno, Hajime Shigemitsu*, Aya Yoshikawa, Yasuko Osakada, Mamoru Fujitsuka, Fumitaka Ishiwari, Akinori Saeki, Kei Ohkubo, Tadashi Mori, and Toshiyuki Kida*Chem. Commun., 2024, 60, 889–892.Development of two-dimensional materials and exploration of their functionalities are significant challenges due to their potential. In this study, we successfully fabricated a supramolecular nanosheet composed of amphiphilic Rose Bengal dyes in an aqueous medium. Furthermore, we elucidated a distinct change in the photosensitisation mechanism induced by nanosheet formation.
@article{bunno2024supramolecular, title = {Supramolecular nanosheet formation-induced photosensitisation mechanism change of Rose Bengal dye in aqueous media}, author = {Bunno, Asuka and Shigemitsu, Hajime and Yoshikawa, Aya and Osakada, Yasuko and Fujitsuka, Mamoru and Ishiwari, Fumitaka and Saeki, Akinori and Ohkubo, Kei and Mori, Tadashi and Kida, Toshiyuki}, journal = {Chem. Commun.}, volume = {60}, number = {7}, pages = {889--892}, year = {2024}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d3cc05731c}, url = {https://doi.org/10.1039/d3cc05731c}, dimensions = {true}, tab = {paper}, } -
 Impact of Symmetry on Chiroptical Properties of Double HelicenesTadashi Mori*Symmetry, 2024, 35, 337–338.
Impact of Symmetry on Chiroptical Properties of Double HelicenesTadashi Mori*Symmetry, 2024, 35, 337–338.The significance of chiral molecules in advanced materials and technologies is undeniable in chemistry yet designing them with desired chiroptical properties lacks reliable strategies.
@article{mori2024impact, title = {Impact of Symmetry on Chiroptical Properties of Double Helicenes}, author = {Mori, Tadashi}, journal = {Symmetry}, volume = {35}, issue = {3}, pages = {337--338}, year = {2024}, publisher = {Symmetry: Culture and Science}, doi = {10.26830/symmetry_2024_3_337}, url = {https://doi.org/10.26830/symmetry_2024_3_337}, dimensions = {true}, tab = {paper}, }
2023
-
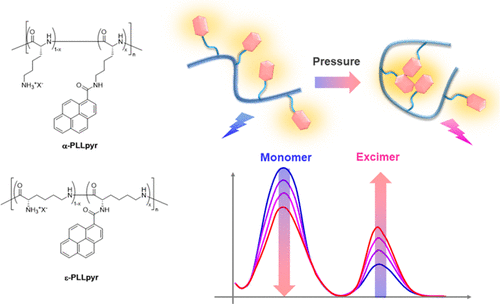 Pressure-responsive polymer chemosensors for hydrostatic-pressure-signal detection: poly-L-lysine–pyrene conjugatesSoshi Wakako, Yumiko Hori, Tomokazu Kinoshita, Takao Saiki, Xinyi Qi, Koki Hasegawa, Yoshitane Imai, Tadashi Mori, Keiichi Nakagawa, and Gaku Fukuhara*ACS Macro Lett., 2023, 12, 1389–1395.
Pressure-responsive polymer chemosensors for hydrostatic-pressure-signal detection: poly-L-lysine–pyrene conjugatesSoshi Wakako, Yumiko Hori, Tomokazu Kinoshita, Takao Saiki, Xinyi Qi, Koki Hasegawa, Yoshitane Imai, Tadashi Mori, Keiichi Nakagawa, and Gaku Fukuhara*ACS Macro Lett., 2023, 12, 1389–1395.Stimulus-responsive polymer materials are an attractive alternative to conventional supramolecular and polymer assemblies for applications in sensing, imaging, and drug-delivery systems. Herein, we synthesized a series of pyrene-labeled α- and ε-poly-l-lysine conjugates with varying degrees of substitution (DSs). Hydrostatic-pressure-UV/vis, fluorescence, and excitation spectroscopies and fluorescence lifetime measurements revealed ground-state conformers and excited-state ensembles emitting fluorescence with variable intensities. The polylysine-based chemosensors demonstrated diverse ratiometric responses to hydrostatic pressure through adjustments in polar solvents, DSs, and polymer backbones. Additionally, the fluorescence chemosensor exhibited a promising glum value of 3.2 × 10–3, indicating potential applications in chiral fluorescent materials. This study offers valuable insights into the development of smart hydrostatic-pressure-responsive polymer materials.
@article{wakako2023pressure, title = {Pressure-responsive polymer chemosensors for hydrostatic-pressure-signal detection: poly-L-lysine--pyrene conjugates}, author = {Wakako, Soshi and Hori, Yumiko and Kinoshita, Tomokazu and Saiki, Takao and Qi, Xinyi and Hasegawa, Koki and Imai, Yoshitane and Mori, Tadashi and Nakagawa, Keiichi and Fukuhara, Gaku}, journal = {ACS Macro Lett.}, volume = {12}, number = {10}, pages = {1389--1395}, year = {2023}, publisher = {ACS Publications}, doi = {10.1021/acsmacrolett.3c00427}, url = {https://doi.org/10.1021/acsmacrolett.3c00427}, dimensions = {true}, tab = {paper}, } -
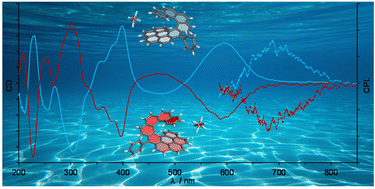 A π-extended phenanthrene-fused aza [7] helicenium as a novel chiroptically-active architecture in organic and aqueous mediaCéline Olivier*, Nao Nagatomo, Tadashi Mori, Nathan McClenaghan, Gediminas Jonusauskas, Brice Kauffmann, Yutaka Kuwahara, Makoto Takafuji, Hirotaka Ihara, and Yann Ferrand*Org. Chemi. Front., 2023, 10, 752–758.
A π-extended phenanthrene-fused aza [7] helicenium as a novel chiroptically-active architecture in organic and aqueous mediaCéline Olivier*, Nao Nagatomo, Tadashi Mori, Nathan McClenaghan, Gediminas Jonusauskas, Brice Kauffmann, Yutaka Kuwahara, Makoto Takafuji, Hirotaka Ihara, and Yann Ferrand*Org. Chemi. Front., 2023, 10, 752–758.The synthesis and characterization of an original π-extended cationic azahelicene is reported. The phenanthrene-fused aza[7]helicene derivative encompasses a total of ten aromatic fused rings leading to a dissymmetric yet helically folded structure, as revealed by NMR and X-ray diffraction analyses. The polyaromatic and cationic nature of the new azahelicenium makes it soluble in both organic and aqueous media, which allowed photophysical studies in solvents of different polarities. The extended chromophoric species demonstrates a broad absorption over the whole visible range and orange-red fluorescence emission. Chiral resolution of the racemate was performed subsequently, affording two optically pure and configurationally stable azahelicenium enantiomers. Multi-band circular dichroism and long-wavelength circularly polarized emission were observed, associated with remarkable absorption and luminescence dissymmetry factors, both in organic and aqueous media.
@article{olivier2023pi, title = {A π-extended phenanthrene-fused aza [7] helicenium as a novel chiroptically-active architecture in organic and aqueous media}, author = {Olivier, Céline and Nagatomo, Nao and Mori, Tadashi and McClenaghan, Nathan and Jonusauskas, Gediminas and Kauffmann, Brice and Kuwahara, Yutaka and Takafuji, Makoto and Ihara, Hirotaka and Ferrand, Yann}, journal = {Org. Chemi. Front.}, volume = {10}, number = {3}, pages = {752--758}, year = {2023}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d2qo01942f}, url = {https://doi.org/10.1039/d2qo01942f}, dimensions = {true}, tab = {paper}, } -
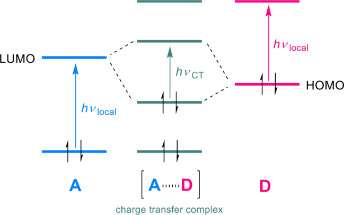 Induction of chirality in the charge transfer complex photochemistryTadashi Mori*J. Photochem. Photobiol., 2023, 17, 100200.
Induction of chirality in the charge transfer complex photochemistryTadashi Mori*J. Photochem. Photobiol., 2023, 17, 100200.Electron donor and acceptor undergo association in solution to form a weak molecular complex, namely a charge transfer complex. The formation of the complex is apparent by its new low-energy absorption band. Upon photoexcitation of this charge transfer band, an intracomplex electron transfer is facilitated to form a contact ion radical pair. The theoretical basis for the complex formation and the subsequent photoinduced electron transfer process has been well established already half a century ago. Nevertheless, the charge transfer complex photochemistry is now in a renaissance, due to its synthetic utility of its radical ion pair. By intentionally sidestepped unproductive back electron transfer process, a desired product is effectively obtained with or without external reagent and/or catalyst. In addition, the photoexcitation of the complex does not require any additional photocatalyst and often conceivable with the visible light. Accordingly, various donor/acceptor pairs and synthetic applications have been emerged as novel photoreaction systems in the last two decades. Some have also addressed the stereoselective transformations. This mini review highlights the recent progress of the asymmetric photoreaction, in particular in synthetic applications, based on the photoexcitation of the charge transfer complex, in comparison with photoredox catalysis.
@article{mori2023induction, title = {Induction of chirality in the charge transfer complex photochemistry}, author = {Mori, Tadashi}, journal = {J. Photochem. Photobiol.}, volume = {17}, pages = {100200}, year = {2023}, publisher = {Elsevier}, doi = {10.1016/j.jpap.2023.100200}, url = {https://doi.org/10.1016/j.jpap.2023.100200}, dimensions = {true}, tab = {paper}, } -
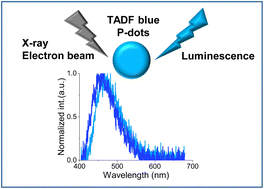 Radioluminescence from polymer dots based on thermally activated delayed fluorescenceDaiki Asanuma, Hieu Thi Minh Nguyen, Zuoyue Liu, Sachiko Tojo, Hajime Shigemitsu, Minoru Yamaji, Kiyohiko Kawai, Tadashi Mori, Toshiyuki Kida, Guillem Pratx*, Mamoru Fujitsuka*, and Yasuko Osakada*Nanoscale Adv., 2023, 5, 3424–3427.
Radioluminescence from polymer dots based on thermally activated delayed fluorescenceDaiki Asanuma, Hieu Thi Minh Nguyen, Zuoyue Liu, Sachiko Tojo, Hajime Shigemitsu, Minoru Yamaji, Kiyohiko Kawai, Tadashi Mori, Toshiyuki Kida, Guillem Pratx*, Mamoru Fujitsuka*, and Yasuko Osakada*Nanoscale Adv., 2023, 5, 3424–3427.We demonstrate that polymer dots doped with thermally activated delayed fluorescence (TADF) molecules clearly exhibit blue radio-luminescence upon hard X-ray and electron beam irradiation, which is a new design for nano-sized scintillators.
@article{asanuma2023radioluminescence, title = {Radioluminescence from polymer dots based on thermally activated delayed fluorescence}, author = {Asanuma, Daiki and Nguyen, Hieu Thi Minh and Liu, Zuoyue and Tojo, Sachiko and Shigemitsu, Hajime and Yamaji, Minoru and Kawai, Kiyohiko and Mori, Tadashi and Kida, Toshiyuki and Pratx, Guillem and Fujitsuka, Mamoru and Osakada, Yasuko}, journal = {Nanoscale Adv.}, volume = {5}, number = {13}, pages = {3424--3427}, year = {2023}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d3na00308f}, url = {https://doi.org/10.1039/d3na00308f}, dimensions = {true}, tab = {paper}, }
2022
-
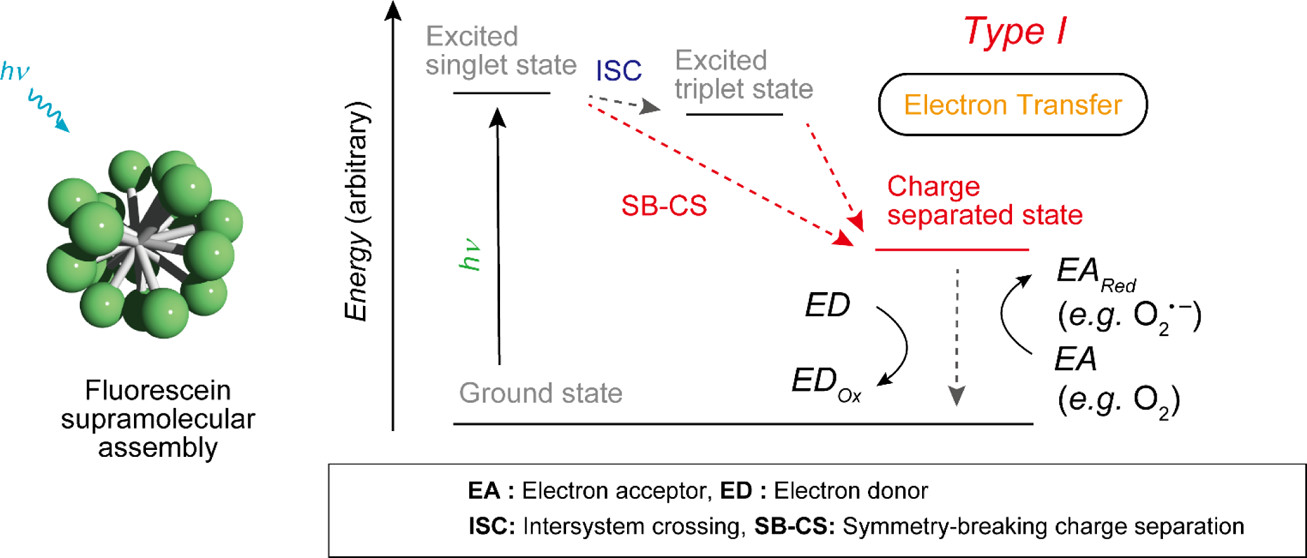 Fluorescein-Based Type I Supramolecular Photosensitizer via Induction of Charge Separation by Self-AssemblyHajime Shigemitsu*, Kei Ohkubo, Kazuhide Sato, Asuka Bunno, Tadashi Mori, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki Kida*JACS Au, 2022, 2, 1472–1478.
Fluorescein-Based Type I Supramolecular Photosensitizer via Induction of Charge Separation by Self-AssemblyHajime Shigemitsu*, Kei Ohkubo, Kazuhide Sato, Asuka Bunno, Tadashi Mori, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki Kida*JACS Au, 2022, 2, 1472–1478.Photosensitizers (PSs) are critical substances with considerable potential for use in non-invasive photomedicine. Type I PSs, which generate reactive radical species by electron transfer from the excited state induced via photoirradiation, attracted much attention because of their suitability for photodynamic therapy (PDT) irrespective of the oxygen concentration. However, most organic PSs are type II, which activates only oxygen, generating singlet oxygen (1O2) via energy transfer from the triplet state. Here, we proposed a strategy to form type I supramolecular PSs (SPSs) utilizing the charge-separated state induced by self-assembly. This was demonstrated using a supramolecular assembly of fluorescein, which is a type II PS in the monomeric state; however, it changes to a type I SPS via self-assembly. The switching mechanism from type II to I via self-assembly was clarified using photophysical and electrochemical analyses, with the type I SPS exhibiting significant PDT effects on cancer cells. This study provides a promising approach for the development of type I PSs based on supramolecular assemblies.
@article{shigemitsu2022fluorescein, title = {Fluorescein-Based Type I Supramolecular Photosensitizer via Induction of Charge Separation by Self-Assembly}, author = {Shigemitsu, Hajime and Ohkubo, Kei and Sato, Kazuhide and Bunno, Asuka and Mori, Tadashi and Osakada, Yasuko and Fujitsuka, Mamoru and Kida, Toshiyuki}, journal = {JACS Au}, volume = {2}, issue = {6}, pages = {1472--1478}, year = {2022}, month = may, publisher = {ACS Publications}, doi = {10.1021/jacsau.2c00243}, url = {https://doi.org/10.1021/jacsau.2c00243}, dimensions = {true}, tab = {paper}, } -
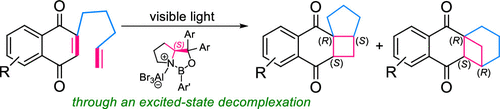 Visible Light-Induced Regio-and Enantiodifferentiating [2+2] Photocycloaddition of 1, 4-Naphthoquinones Mediated by Oppositely Coordinating 1, 3, 2-Oxazaborolidine Chiral Lewis AcidNao Shimizu, Hajime Shigemitsu, Toshiyuki Kida, Thorsten Bach, and Tadashi Mori*J. Org. Chem., 2022, 87, 8071–8083.
Visible Light-Induced Regio-and Enantiodifferentiating [2+2] Photocycloaddition of 1, 4-Naphthoquinones Mediated by Oppositely Coordinating 1, 3, 2-Oxazaborolidine Chiral Lewis AcidNao Shimizu, Hajime Shigemitsu, Toshiyuki Kida, Thorsten Bach, and Tadashi Mori*J. Org. Chem., 2022, 87, 8071–8083.A range of asymmetric photochemical transformations using visible light have recently become considerably attractive. Among the various approaches, chiral Lewis acid association to enones for [2 + 2] and ortho photocycloadditions and oxadi-π-methane rearrangements have shown to be very promising. Naturally, chiral Lewis acid coordination protects one of the prochiral faces of the C═C double bond, which enables an effective enantiodifferentiation in the following bond-forming process(es). Here, we studied regio- and enantiodifferentiating [2 + 2] photocycloaddition reactions of naphthoquinone derivatives mediated by chiral oxazaborolidines. A stereochemical control was quite challenging for the 2-ene-1,4-dione substrate, as a double coordination of Lewis acid essentially cancels out the face selectivity, and a mono-coordination to each carbonyl group leads to an opposite stereochemical outcome. Furthermore, a stepwise coordination in the ground state of Lewis acid in a 1:1 fashion was practically inaccessible. We found that the excited-state decomplexation is a key to accomplish high regio- and enantioselectivities in the photocycloaddition of an ene-dione.
@article{shimizu2022visible, title = {Visible Light-Induced Regio-and Enantiodifferentiating [2+2] Photocycloaddition of 1, 4-Naphthoquinones Mediated by Oppositely Coordinating 1, 3, 2-Oxazaborolidine Chiral Lewis Acid}, author = {Shimizu, Nao and Shigemitsu, Hajime and Kida, Toshiyuki and Bach, Thorsten and Mori, Tadashi}, journal = {J. Org. Chem.}, volume = {87}, issue = {12}, pages = {8071--8083}, year = {2022}, publisher = {ACS Publications}, doi = {10.1021/acs.joc.2c00730}, url = {https://doi.org/10.1021/acs.joc.2c00730}, dimensions = {true}, tab = {paper}, } -
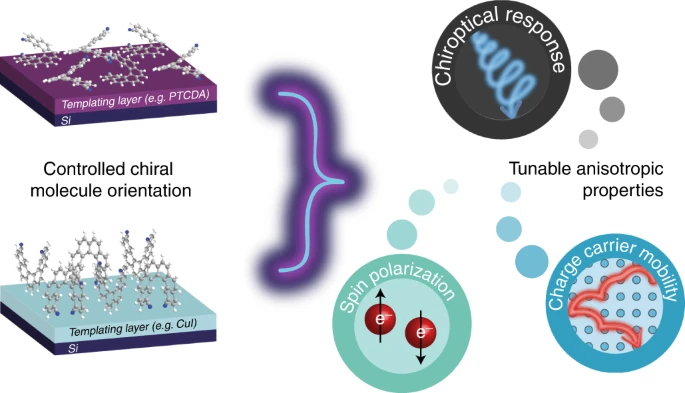 Controlling anisotropic properties by manipulating the orientation of chiral small moleculesJessica Wade*, Francesco Salerno, Rachel C Kilbride, Dong Kuk Kim, Julia A Schmidt, Joel A Smith, Luc M LeBlanc, Emma H Wolpert, Adebayo A Adeleke, Erin R Johnson, Jenny Nelson, Tadashi Mori, Kim E Jelfs, Sandrine Heutz, and Matthew J Fuchter*Nat. Chem., 2022, 14, 1383–1389.
Controlling anisotropic properties by manipulating the orientation of chiral small moleculesJessica Wade*, Francesco Salerno, Rachel C Kilbride, Dong Kuk Kim, Julia A Schmidt, Joel A Smith, Luc M LeBlanc, Emma H Wolpert, Adebayo A Adeleke, Erin R Johnson, Jenny Nelson, Tadashi Mori, Kim E Jelfs, Sandrine Heutz, and Matthew J Fuchter*Nat. Chem., 2022, 14, 1383–1389.Chiral π-conjugated molecules bring new functionality to technological applications and represent an exciting, rapidly expanding area of research. Their functional properties, such as the absorption and emission of circularly polarized light or the transport of spin-polarized electrons, are highly anisotropic. As a result, the orientation of chiral molecules critically determines the functionality and efficiency of chiral devices. Here we present a strategy to control the orientation of a small chiral molecule (2,2′-dicyano[6]helicene) by the use of organic and inorganic templating layers. Such templating layers can either force 2,2′-dicyano[6]helicene to adopt a face-on orientation and self-assemble into upright supramolecular columns oriented with their helical axis perpendicular to the substrate, or an edge-on orientation with parallel-lying supramolecular columns. Through such control, we show that low- and high-energy chiroptical responses can be independently ‘turned on’ or ‘turned off’. The templating methodologies described here provide a simple way to engineer orientational control and, by association, anisotropic functional properties of chiral molecular systems for a range of emerging technologies.
@article{wade2022controlling, title = {Controlling anisotropic properties by manipulating the orientation of chiral small molecules}, author = {Wade, Jessica and Salerno, Francesco and Kilbride, Rachel C and Kim, Dong Kuk and Schmidt, Julia A and Smith, Joel A and LeBlanc, Luc M and Wolpert, Emma H and Adeleke, Adebayo A and Johnson, Erin R and Nelson, Jenny and Mori, Tadashi and Jelfs, Kim E and Heutz, Sandrine and Fuchter, Matthew J}, journal = {Nat. Chem.}, volume = {14}, number = {12}, pages = {1383--1389}, year = {2022}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41557-022-01044-6}, url = {https://doi.org/10.1038/s41557-022-01044-6}, dimensions = {true}, tab = {paper}, } -
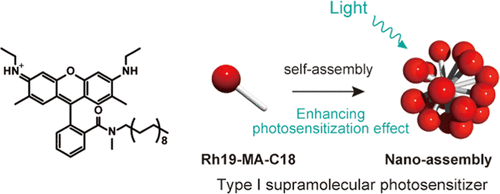 Amphiphilic rhodamine nano-assembly as a type I supramolecular photosensitizer for photodynamic therapyHajime Shigemitsu*, Kazuhide Sato, Satomi Hagio, Youhei Tani, Tadashi Mori, Kei Ohkubo, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki Kida*ACS Appl. Nano Mater., 2022, 5, 14954–14960.
Amphiphilic rhodamine nano-assembly as a type I supramolecular photosensitizer for photodynamic therapyHajime Shigemitsu*, Kazuhide Sato, Satomi Hagio, Youhei Tani, Tadashi Mori, Kei Ohkubo, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki Kida*ACS Appl. Nano Mater., 2022, 5, 14954–14960.Photodynamic therapy (PDT) is a promising clinical method for treating a wide range of cancers. Recently, PDT employing type I photosensitizers (PSs) has attracted considerable attention owing to the feasibility of efficient PDT under hypoxic conditions. Particularly, type I supramolecular PSs (SPSs) are promising candidates owing to their functional extensibility by facile hybridization. However, type I SPSs are rare, and the development strategy has not been established yet. In this work, we demonstrated that supramolecular assembly of a simple amphiphilic rhodamine 19 (Rh19-MA-C18) functioned as a type I SPS and exhibited significant PDT effect on cancer cells and cancer-bearing mice. This work demonstrates the potential of supramolecular nano-assembly composed of an amphiphilic rhodamine dye as an efficient type I SPS.
@article{shigemitsu2022amphiphilic, title = {Amphiphilic rhodamine nano-assembly as a type I supramolecular photosensitizer for photodynamic therapy}, author = {Shigemitsu, Hajime and Sato, Kazuhide and Hagio, Satomi and Tani, Youhei and Mori, Tadashi and Ohkubo, Kei and Osakada, Yasuko and Fujitsuka, Mamoru and Kida, Toshiyuki}, journal = {ACS Appl. Nano Mater.}, volume = {5}, number = {10}, pages = {14954--14960}, year = {2022}, publisher = {ACS Publications}, doi = {10.1021/acsanm.2c03192}, url = {https://doi.org/10.1021/acsanm.2c03192}, dimensions = {true}, tab = {paper}, } -
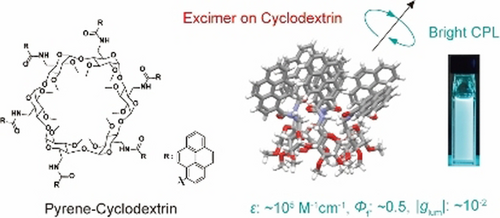 Cyclodextrins with multiple pyrenyl groups: an approach to organic molecules exhibiting bright excimer circularly polarized luminescenceHajime Shigemitsu*, Kosei Kawakami, Yuuya Nagata, Rikuo Kajiwara, Shintaro Yamada, Tadashi Mori, and Toshiyuki Kida*Angew. Chem. Int. Ed., 2022, 134, e202114700.
Cyclodextrins with multiple pyrenyl groups: an approach to organic molecules exhibiting bright excimer circularly polarized luminescenceHajime Shigemitsu*, Kosei Kawakami, Yuuya Nagata, Rikuo Kajiwara, Shintaro Yamada, Tadashi Mori, and Toshiyuki Kida*Angew. Chem. Int. Ed., 2022, 134, e202114700.We report a simple and effective approach to organic molecules exhibiting bright circularly polarized luminescence (CPL) by combining a chiral cyclic molecular scaffold and multiple excimer-enabling moieties. An α-cyclodextrin (CyD) scaffold was modified with six pyrenyl groups to obtain pyrene–cyclodextrins (PCDs) in a one-step synthesis from commercially available compounds. The PCDs exhibited high molar extinction coefficients (ϵ≈105 M−1 cm−1), polarized emission with a good dissymmetry factor (|glum|≈10−2), and quantum yield (Φf≈0.5). Owing to the excellent photophysical properties of the PCDs, the circularly polarized luminescence brightness (BCPL) reached 340 M−1 cm−1. Photophysical and chiroptical studies of the PCDs with only five pyrene units and with linkers of various lengths connecting the CyD with the pyrene units revealed that the formation of a pyrene excimer in a spatially crowded environment is crucial for CPL anisotropy. This study paves the way for the development of bright CPL organic molecules.
@article{shigemitsu2022cyclodextrins, title = {Cyclodextrins with multiple pyrenyl groups: an approach to organic molecules exhibiting bright excimer circularly polarized luminescence}, author = {Shigemitsu, Hajime and Kawakami, Kosei and Nagata, Yuuya and Kajiwara, Rikuo and Yamada, Shintaro and Mori, Tadashi and Kida, Toshiyuki}, journal = {Angew. Chem. Int. Ed.}, volume = {134}, number = {8}, pages = {e202114700}, year = {2022}, publisher = {Wiley Online Library}, doi = {10.1002/anie.202114700}, url = {https://doi.org/10.1002/anie.202114700}, dimensions = {true}, tab = {paper}, } -
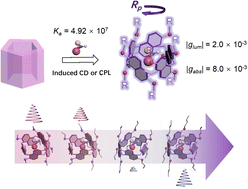 Chiroptical induction with prism [5] arene alkoxy-homologsXiaotong Liang, Yanling Shen, Dayang Zhou, Jiecheng Ji, Hongtao Wang, Ting Zhao, Tadashi Mori, Wanhua Wu, and Cheng YangChem. Commun., 2022, 58, 13584–13587.
Chiroptical induction with prism [5] arene alkoxy-homologsXiaotong Liang, Yanling Shen, Dayang Zhou, Jiecheng Ji, Hongtao Wang, Ting Zhao, Tadashi Mori, Wanhua Wu, and Cheng YangChem. Commun., 2022, 58, 13584–13587.The complexation of prism[5]arenes with amino acid derivatives showed association constants of up to 107 M−1, significant CD with gabs of up to 0.8 × 10−2 and CPL with glum of 2 × 10−3. The absolute configuration-CD signal correlation was established. The CD spectra varied significantly with the substituents on the prism[5]arenes.
@article{liang2022chiroptical, title = {Chiroptical induction with prism [5] arene alkoxy-homologs}, author = {Liang, Xiaotong and Shen, Yanling and Zhou, Dayang and Ji, Jiecheng and Wang, Hongtao and Zhao, Ting and Mori, Tadashi and Wu, Wanhua and Yang, Cheng}, journal = {Chem. Commun.}, volume = {58}, number = {98}, pages = {13584--13587}, year = {2022}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d2cc05690a}, url = {https://doi.org/10.1039/d2cc05690a}, dimensions = {true}, tab = {paper}, } -
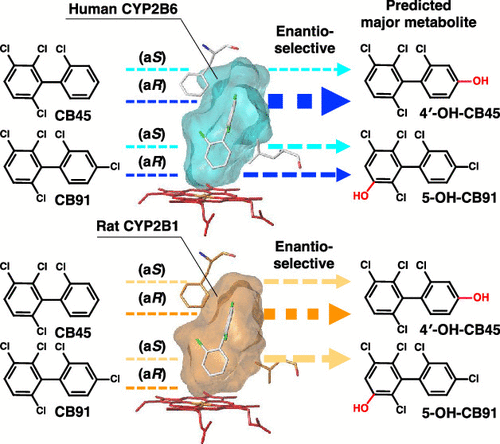 Differences in enantioselective hydroxylation of 2, 2’, 3, 6-tetrachlorobiphenyl (CB45) and 2, 2’, 3, 4’, 6-pentachlorobiphenyl (CB91) by human and rat CYP2B subfamiliesHideyuki Inui*, Terushi Ito, Chiharu Miwa, Yuki Haga, Makoto Kubo, Toshimasa Itoh, Keiko Yamamoto, Masayuki Miyaoka, Tadashi Mori, Harunobu Tsuzuki, and othersEnviron. Sci. Technol., 2022, 56, 10204–10215.
Differences in enantioselective hydroxylation of 2, 2’, 3, 6-tetrachlorobiphenyl (CB45) and 2, 2’, 3, 4’, 6-pentachlorobiphenyl (CB91) by human and rat CYP2B subfamiliesHideyuki Inui*, Terushi Ito, Chiharu Miwa, Yuki Haga, Makoto Kubo, Toshimasa Itoh, Keiko Yamamoto, Masayuki Miyaoka, Tadashi Mori, Harunobu Tsuzuki, and othersEnviron. Sci. Technol., 2022, 56, 10204–10215.Although polychlorinated biphenyls (PCBs) were commercially banned half a century ago, contamination of the environment and organisms by PCBs is still observed. PCBs show high persistence and bioaccumulation, resulting in toxicity. Among PCBs, chiral PCBs with more than three chlorine atoms at the ortho-position exhibit developmental and neurodevelopmental toxicity. Because toxicity is dependent on the atropisomer, atropisomer-specific metabolism is vital in determining toxicity. However, structural information on enantioselective metabolism remains elusive. Cytochrome P450 (CYP, P450) monooxygenases, particularly human CYP2B6 and rat CYP2B1, metabolize separated atropisomers of 2,2’,3,6-tetrachlorobiphenyl (CB45) and 2,2’,3,4’,6-pentachlorobiphenyl (CB91) to dechlorinated and hydroxylated metabolites. Docking studies using human CYP2B6 predict 4′-hydroxy (OH)-CB45 from (aR)-CB45 as a major metabolite of CB45. Di-OH- and dechlorinated OH-metabolites from human CYP2B6 and rat CYP2B1 are also detected. Several hydroxylated metabolites are derived from CB91 by both P450s; 5-OH-CB91 is predicted as a major metabolite. CB91 dechlorination is also detected by identifying 3-OH-CB51. A stable conformation of PCBs in the substrate-binding cavity and close distance to P450 heme are responsible for high metabolizing activities. As hydroxylation and dechlorination change PCB toxicity, this approach helps understand the possible toxicity of chiral PCBs in mammals.
@article{inui2022differences, title = {Differences in enantioselective hydroxylation of 2, 2', 3, 6-tetrachlorobiphenyl (CB45) and 2, 2', 3, 4', 6-pentachlorobiphenyl (CB91) by human and rat CYP2B subfamilies}, author = {Inui, Hideyuki and Ito, Terushi and Miwa, Chiharu and Haga, Yuki and Kubo, Makoto and Itoh, Toshimasa and Yamamoto, Keiko and Miyaoka, Masayuki and Mori, Tadashi and Tsuzuki, Harunobu and others}, journal = {Environ. Sci. Technol.}, volume = {56}, number = {14}, pages = {10204--10215}, year = {2022}, publisher = {ACS Publications}, doi = {10.1021/acs.est.2c01155}, url = {https://doi.org/10.1021/acs.est.2c01155}, dimensions = {true}, tab = {paper}, } - Photochemistry Controlled by Weak Supramolecular InteractionsTadashi Mori*Manufacturing and Technology, 2022, 74, 65–68.
@article{mori2022photochemistry, title = {Photochemistry Controlled by Weak Supramolecular Interactions}, author = {Mori, Tadashi}, journal = {Manufacturing and Technology}, volume = {74}, issue = {1}, pages = {65--68}, year = {2022} }
2021
-
 Overtemperature-protection intelligent molecular chiroptical photoswitchesJiabin Yao, Wanhua Wu*, Chao Xiao, Dan Su, Zhihui Zhong, Tadashi Mori, and Cheng Yang*Nat. Commun., 2021, 12, 2600.
Overtemperature-protection intelligent molecular chiroptical photoswitchesJiabin Yao, Wanhua Wu*, Chao Xiao, Dan Su, Zhihui Zhong, Tadashi Mori, and Cheng Yang*Nat. Commun., 2021, 12, 2600.Stimuli-responsive intelligent molecular machines/devices are of current research interest due to their potential application in minimized devices. Constructing molecular machines/devices capable of accomplishing complex missions is challenging, demanding coalescence of various functions into one molecule. Here we report the construction of intelligent molecular chiroptical photoswitches based on azobenzene-fused bicyclic pillar[n]arene derivatives, which we defined as molecular universal joints (MUJs). The Z/E photoisomerization of the azobenzene moiety of MUJs induces rolling in/out conformational switching of the azobenzene-bearing side-ring and consequently leads to planar chirality switching of MUJs. Meanwhile, temperature variation was demonstrated to also cause conformational/chiroptical inversion due to the significant entropy change during the ring-flipping. As a result, photo-induced chiroptical switching could be prohibited when the temperature exceeded an upper limit, demonstrating an intelligent molecular photoswitch having over-temperature protection function, which is in stark contrast to the low-temperature-gating effect commonly encountered.
@article{yao2021overtemperature, title = {Overtemperature-protection intelligent molecular chiroptical photoswitches}, author = {Yao, Jiabin and Wu, Wanhua and Xiao, Chao and Su, Dan and Zhong, Zhihui and Mori, Tadashi and Yang, Cheng}, journal = {Nat. Commun.}, volume = {12}, issue = {1}, pages = {2600}, year = {2021}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41467-021-22880-z}, url = {https://doi.org/10.1038/s41467-021-22880-z}, dimensions = {true}, tab = {paper}, } -
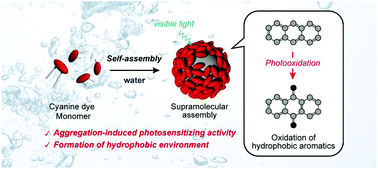 A cyanine dye based supramolecular photosensitizer enabling visible-light-driven organic reaction in waterHajime Shigemitsu*, Tomoe Tamemoto, Kei Ohkubo, Tadashi Mori, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki KidaChem. Commun., 2021, 57, 11217–11220.
A cyanine dye based supramolecular photosensitizer enabling visible-light-driven organic reaction in waterHajime Shigemitsu*, Tomoe Tamemoto, Kei Ohkubo, Tadashi Mori, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki KidaChem. Commun., 2021, 57, 11217–11220.We report the aggregation-induced photosensitizing activity of a cyanine dye in water and the mechanism. In addition, using the supramolecular assembly, visible-light-driven photooxidation of hydrophobic aromatic compounds in water was successfully performed.
@article{shigemitsu2021cyanine, title = {A cyanine dye based supramolecular photosensitizer enabling visible-light-driven organic reaction in water}, author = {Shigemitsu, Hajime and Tamemoto, Tomoe and Ohkubo, Kei and Mori, Tadashi and Osakada, Yasuko and Fujitsuka, Mamoru and Kida, Toshiyuki}, journal = {Chem. Commun.}, volume = {57}, issue = {85}, pages = {11217--11220}, year = {2021}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d1cc04685c}, url = {https://doi.org/10.1039/d1cc04685c}, dimensions = {true}, tab = {paper}, } -
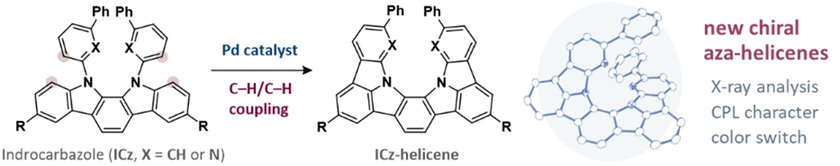 Synthesis, Structure, and Chiroptical Properties of Indolo-and Pyridopyrrolo-Carbazole-Based C2-Symmetric AzahelicenesTaisei Taniguchi, Yuji Nishii, Tadashi Mori*, Ken-ichi Nakayama, and Masahiro Miura*Chem. Eur. J., 2021, 27, 7356–7361.
Synthesis, Structure, and Chiroptical Properties of Indolo-and Pyridopyrrolo-Carbazole-Based C2-Symmetric AzahelicenesTaisei Taniguchi, Yuji Nishii, Tadashi Mori*, Ken-ichi Nakayama, and Masahiro Miura*Chem. Eur. J., 2021, 27, 7356–7361.Treatment of 11,12-bis(1,1’-biphenyl-3-yl or 6-phenylpyridin-2-yl)-substituted 11,12-dihydro-indolo[2,3-a]carbazole with an oxidizing system of Pd(II)/Ag(I) induced effective double dehydrogenative cyclization to afford the corresponding π-extended azahelicenes. The optical resolutions were readily achieved by a preparative chiral HPLC. It was found that the pyridopyrrolo-carbazole-based azahelicene that contains four nitrogen atoms exhibits ca. 6 times larger dissymmetry factors both in circularly dichroism (CD) and circularly polarized luminescence (CPL), |gCD| and |gCPL| values being 1.1×10−2 and 4.4×10−3, respectively, as compared with the parent indolocarbazole-based azahelicene. Theoretical calculations at the RI-CC2 level were employed to rationalize the observed enhanced chiroptical responses. The (chir)optical properties of the former helicene was further tuned by a protonation leading to remarkable red-shift with a considerable enhancement of the |gCPL| value.
@article{taniguchi2021synthesis, title = {Synthesis, Structure, and Chiroptical Properties of Indolo-and Pyridopyrrolo-Carbazole-Based C2-Symmetric Azahelicenes}, author = {Taniguchi, Taisei and Nishii, Yuji and Mori, Tadashi and Nakayama, Ken-ichi and Miura, Masahiro}, journal = {Chem. Eur. J.}, volume = {27}, issue = {26}, pages = {7356--7361}, year = {2021}, publisher = {Wiley Online Library}, doi = {10.1002/chem.202100327}, url = {https://doi.org/10.1002/chem.202100327}, dimensions = {true}, tab = {paper}, } -
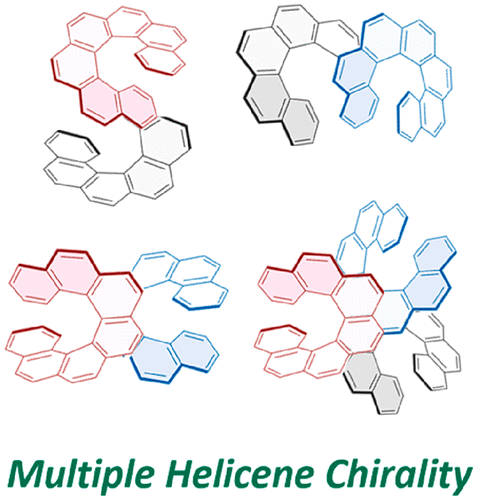 Chiroptical properties of symmetric double, triple, and multiple helicenesTadashi Mori*Chem. Rev., 2021, 121, 2373–2412.
Chiroptical properties of symmetric double, triple, and multiple helicenesTadashi Mori*Chem. Rev., 2021, 121, 2373–2412.Helicenes have attracted considerable attention due to their inherent helical chirality and extended π-conjugation. Recently, rapid progress has been witnessed in the preparation of double, triple, quadruple, quintuple, and sextuple helicenes, where plural helicene moieties are symmetrically arranged in a single molecule. While synthetic efforts and X-ray crystallographic analyses devoted to these multiple helicenes and theoretical investigations on their isomerization and racemization behaviors have been relatively well documented and reviewed in the literature, the chiroptical properties of the multiple helicities have been somewhat overlooked. This review discourses the cumulative and systematic investigations on the chiroptical properties such as the circular dichroism (CD) and circularly polarized luminescence (CPL) of multiple helicenes. Although the number and structural variations of multiple helicenes reported to date have been fairly limited, this review overviews the current status of the chemistry of multiple helicenes from the viewpoint of chiroptical properties and provides insights into the design principle for advanced chiroptical materials through the proper arrangement of multiple helices, highlighting the impact of the molecular symmetry on the chiroptical responses.
@article{mori2021chiroptical, title = {Chiroptical properties of symmetric double, triple, and multiple helicenes}, author = {Mori, Tadashi}, journal = {Chem. Rev.}, volume = {121}, issue = {4}, pages = {2373--2412}, year = {2021}, publisher = {ACS Publications}, doi = {10.1021/acs.chemrev.0c01017}, url = {https://doi.org/10.1021/acs.chemrev.0c01017}, dimensions = {true}, tab = {review}, }
2020
-
 Enhancing Photostability of a Coumarin Dye by Self-inclusion into a Cyclodextrin Cavity in Aqueous Solution and Living CellsHajime Shigemitsu*, Keigo Matsuda, Tadashi Mori, Hirotaka Nakatsuji, Michiya Matsusaki, and Toshiyuki Kida*Asian J. Org. Chem., 2020, 9, 2112–2115.
Enhancing Photostability of a Coumarin Dye by Self-inclusion into a Cyclodextrin Cavity in Aqueous Solution and Living CellsHajime Shigemitsu*, Keigo Matsuda, Tadashi Mori, Hirotaka Nakatsuji, Michiya Matsusaki, and Toshiyuki Kida*Asian J. Org. Chem., 2020, 9, 2112–2115.Photostable organic dyes are highly demanded for longtime or super-resolution bioimaging. Herein, we demonstrate effective improvement of photostability of a coumarin dye (Pacific Blue (PB)) by steric protection using a cyclodextrin. The PB conjugated cyclodextrin (PB−CD) showed 2.8 times higher photostability than PB ethyl amide (PB−EA) which is a comparative compound in vitro. The cyclodextrin conjugation to PB not only protect PB unit from reactive chemicals, but also suppress reactive oxygen species (ROS) generation which causes dye degradation. The effects afford significant improvement of photostability of PB dye. Finally, the photostability of PB−CD was evaluated using confocal laser scanning microscopy (CLSM) and the significant improvement effect was proofed even in living cells.
@article{shigemitsu2020enhancing, title = {Enhancing Photostability of a Coumarin Dye by Self-inclusion into a Cyclodextrin Cavity in Aqueous Solution and Living Cells}, author = {Shigemitsu, Hajime and Matsuda, Keigo and Mori, Tadashi and Nakatsuji, Hirotaka and Matsusaki, Michiya and Kida, Toshiyuki}, journal = {Asian J. Org. Chem.}, volume = {9}, issue = {12}, pages = {2112--2115}, year = {2020}, publisher = {Wiley Online Library}, doi = {10.1002/ajoc.202000365}, url = {https://doi.org/10.1002/ajoc.202000365}, dimensions = {true}, tab = {paper}, } -
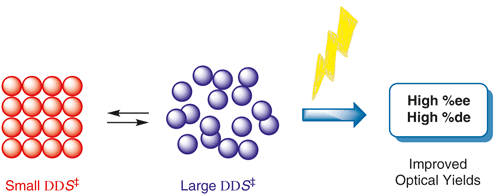 Relevance of the entropy factor in stereoselectivity control of asymmetric photoreactionsTadashi Mori*Synlett, 2020, 31, 1259–1267.
Relevance of the entropy factor in stereoselectivity control of asymmetric photoreactionsTadashi Mori*Synlett, 2020, 31, 1259–1267.Entropy as well as enthalpy factors play substantial roles in various chemical phenomena such as equilibrium and reactions. However, the entropy factors are frequently underestimated in most instances, particularly in synthetic chemistry. In reality, the entropy factor can be in competition with the enthalpy factor or can even be decisive in determining the overall free or activation energy change upon molecular interaction and chemical transformation, particularly where weak interactions in ground and/or excited states are significant. In this account, we overview the importance of the entropy factor in various chemical phenomena in both thermodynamics and kinetics and in the ground and excited states. It is immediately apparent that many diastereo- and enantioselective photoreactions are entropy-controlled. Recent advances on the entropy-control concept in asymmetric photoreactions are further discussed. Understanding the entropy-control concept will pave the way to improve, fine-tune, and even invert the chemo- and stereoselectivity of relevant chemical phenomena.
@article{mori2020relevance, title = {Relevance of the entropy factor in stereoselectivity control of asymmetric photoreactions}, author = {Mori, Tadashi}, journal = {Synlett}, volume = {31}, issue = {13}, pages = {1259--1267}, year = {2020}, publisher = {Georg Thieme Verlag}, doi = {10.1055/s-0040-1707962}, url = {https://doi.org/10.1055/s-0040-1707962}, dimensions = {true}, tab = {review}, } -
 Enantiodifferentiating Photodimerization of a 2, 6-Disubstituted Anthracene Assisted by Supramolecular Double-Helix Formation with Chiral AminesAkio Urushima, Daisuke Taura, Makoto Tanaka, Naomichi Horimoto, Junki Tanabe, Naoki Ousaka, Tadashi Mori, and Eiji Yashima*Angew. Chem. Int. Ed., 2020, 59, 7478–7486.
Enantiodifferentiating Photodimerization of a 2, 6-Disubstituted Anthracene Assisted by Supramolecular Double-Helix Formation with Chiral AminesAkio Urushima, Daisuke Taura, Makoto Tanaka, Naomichi Horimoto, Junki Tanabe, Naoki Ousaka, Tadashi Mori, and Eiji Yashima*Angew. Chem. Int. Ed., 2020, 59, 7478–7486.A novel 2,6-anthrylene-linked bis(m-terphenylcarboxylic acid) strand (1) self-associates into a racemic double-helix. In the presence of chiral mono- and diamines, either a right- or left-handed double-helix was predominantly induced by chiral amines sandwiched between the carboxylic acid strands with accompanying stacking of the two prochiral anthracene linker units in an enantiotopic face-selective way, as revealed by circular dichroism and NMR spectral analyses. The photoirradiation of the optically active double helices complexed with chiral amines proceeded in a diastereo- (anti or syn) and enantiodifferentiating way to afford the chiral anti-photodimer with up to 98% enantiomeric excess when (R)-phenylethylamine was used as a chiral double-helix inducer. The resulting optically active anti-photodimer can recognize the chirality of amines and diastereoselectively complex with chiral amines.
@article{urushima2020enantiodifferentiating, title = {Enantiodifferentiating Photodimerization of a 2, 6-Disubstituted Anthracene Assisted by Supramolecular Double-Helix Formation with Chiral Amines}, author = {Urushima, Akio and Taura, Daisuke and Tanaka, Makoto and Horimoto, Naomichi and Tanabe, Junki and Ousaka, Naoki and Mori, Tadashi and Yashima, Eiji}, journal = {Angew. Chem. Int. Ed.}, volume = {59}, issue = {19}, pages = {7478--7486}, year = {2020}, publisher = {Wiley Online Library}, doi = {10.1002/anie.201916103}, url = {https://doi.org/10.1002/anie.201916103}, dimensions = {true}, tab = {paper}, } -
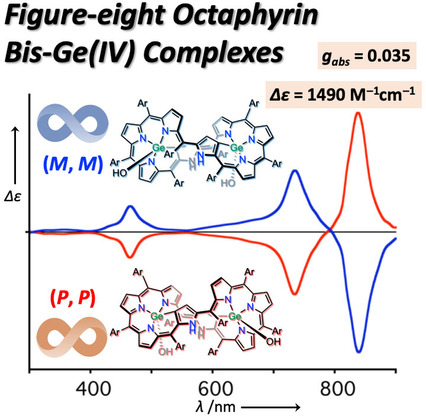 Figure-eight Octaphyrin Bis-Ge (IV) Complexes: Synthesis, Structures, Aromaticity, and Chiroptical PropertiesMondo Izawa, Taisuke Suito, Shin-ichiro Ishida, Daiki Shimizu, Takayuki Tanaka*, Tadashi Mori*, and Atsuhiro Osuka*Chem. Asian J., 2020, 15, 1440–1448.
Figure-eight Octaphyrin Bis-Ge (IV) Complexes: Synthesis, Structures, Aromaticity, and Chiroptical PropertiesMondo Izawa, Taisuke Suito, Shin-ichiro Ishida, Daiki Shimizu, Takayuki Tanaka*, Tadashi Mori*, and Atsuhiro Osuka*Chem. Asian J., 2020, 15, 1440–1448.Highly twisted structures of expanded porphyrin provide a prominent basis to unravel the relationship between aromaticity and chirality. Here we report the synthesis of bis-Ge(IV) complexes of [38]octaphyrin that display rigid figure-eight structures. Two bis-Ge(IV) [38]octaphyrin isomers with respect to the stereochemistry of the axial hydroxy groups on the germanium ions were obtained and found to be aromatic. Upon oxidation with MnO2, these [38]octaphyrin complexes were converted to a single syn-type isomer of [36]octaphyrin with retained figure-eight conformation. The enantiomers have been successfully separated by HPLC equipped with a chiral stationary phase. While aromatic [38]octaphyrin Ge(IV) complexes showed quite large molar circular dichroism of up to Δϵ=1500 M−1cm−1 with a dissymmetry factor gabs of 0.035, weakly antiaromatic [36]octaphyrin Ge(IV) complexes underscored moderate values; Δϵ=540 M−1cm−1 with gabs of 0.023. Thus, the figure-eight octaphyrin scaffold has been proved to be an attractive platform for novel chiroptical materials with tunable aromaticity.
@article{izawa2020figure, title = {Figure-eight Octaphyrin Bis-Ge (IV) Complexes: Synthesis, Structures, Aromaticity, and Chiroptical Properties}, author = {Izawa, Mondo and Suito, Taisuke and Ishida, Shin-ichiro and Shimizu, Daiki and Tanaka, Takayuki and Mori, Tadashi and Osuka, Atsuhiro}, journal = {Chem. Asian J.}, volume = {15}, issue = {9}, pages = {1440--1448}, year = {2020}, publisher = {Wiley Online Library}, doi = {10.1002/asia.202000159}, url = {https://doi.org/10.1002/asia.202000159}, dimensions = {true}, tab = {paper}, } -
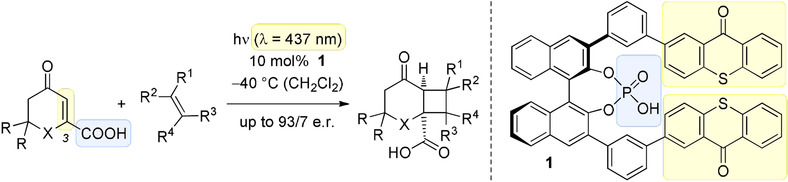 A thioxanthone sensitizer with a chiral phosphoric acid binding site: properties and applications in visible light-mediated cycloadditionsFranziska Pecho, You-Quan Zou, Johannes Gramüller, Tadashi Mori, Stefan M Huber, Andreas Bauer, Ruth M Gschwind, and Thorsten Bach*Chem. Eur. J., 2020, 26, 5190–5194.
A thioxanthone sensitizer with a chiral phosphoric acid binding site: properties and applications in visible light-mediated cycloadditionsFranziska Pecho, You-Quan Zou, Johannes Gramüller, Tadashi Mori, Stefan M Huber, Andreas Bauer, Ruth M Gschwind, and Thorsten Bach*Chem. Eur. J., 2020, 26, 5190–5194.A chiral phosphoric acid with a 2,2’-binaphthol core was prepared that displays two thioxanthone moieties at the 3,3’-position as light-harvesting antennas. Despite its relatively low triplet energy, the phosphoric acid was found to be an efficient catalyst for the enantioselective intermolecular [2+2] photocycloaddition of β-carboxyl-substituted cyclic enones (e.r. up to 93:7). Binding of the carboxylic acid to the sensitizer is suggested by NMR studies and by DFT calculations to occur by means of two hydrogen bonds. The binding event not only enables an enantioface differentiation but also modulates the triplet energy of the substrates.
@article{pecho2020thioxanthone, title = {A thioxanthone sensitizer with a chiral phosphoric acid binding site: properties and applications in visible light-mediated cycloadditions}, author = {Pecho, Franziska and Zou, You-Quan and Gramüller, Johannes and Mori, Tadashi and Huber, Stefan M and Bauer, Andreas and Gschwind, Ruth M and Bach, Thorsten}, journal = {Chem. Eur. J.}, volume = {26}, issue = {23}, pages = {5190--5194}, year = {2020}, publisher = {Wiley Online Library}, doi = {10.1002/chem.202000720}, url = {https://doi.org/10.1002/chem.202000720}, dimensions = {true}, tab = {paper}, } -
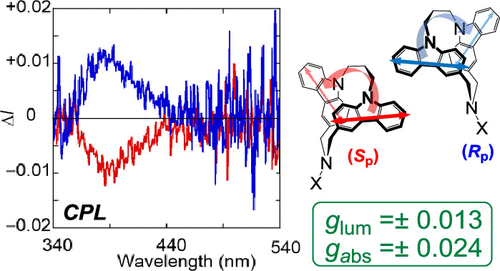 Combined Experimental and Theoretical Studies on Planar Chirality of Partially Overlapped C2-Symmetric [3.3](3,9)DicarbazolophanesKeita Tani*, Kanae Imafuku, Miyuki Eiraku Masaki, Haruka Kato, Kazushige Hori, Koji Kubono, Masatsugu Taneda, Takunori Harada, Kenta Goto, Fumito Tani, and Tadashi Mori*J. Phys. Chem. A, 2020, 124, 2057–2063.
Combined Experimental and Theoretical Studies on Planar Chirality of Partially Overlapped C2-Symmetric [3.3](3,9)DicarbazolophanesKeita Tani*, Kanae Imafuku, Miyuki Eiraku Masaki, Haruka Kato, Kazushige Hori, Koji Kubono, Masatsugu Taneda, Takunori Harada, Kenta Goto, Fumito Tani, and Tadashi Mori*J. Phys. Chem. A, 2020, 124, 2057–2063.Partially overlapped dicarbazolophanes exhibit a planar chirality. In this study, C2-symmetrical [3.3](3,9)dicarbazolophane derivatives (CZ1–CZ3) have been optically resolved by preparative chiral high-performance liquid chromatography for the first time. In their circular dichroism (CD) spectra, moderate Cotton effects (CEs) were observed for their 1Lb and 1La transitions (|Δε| = 10–12 and 51–57 M–1 cm–1, respectively), while intense CEs were notified in their 1B transitions (|Δε| = 156–216 M–1 cm–1), absorption dissymmetry (gabs) factors being in orders of 10–2. Circularly polarized luminescence spectrum was also obtained for cyanamide derivative CZ1, with a comparative luminescence dissymmetry (glum) factor of 0.013. A computational investigation was applied to address the factors for such remarkable chiroptical responses in these dicarbazolophanes of planar chirality. Absolute configurations were unambiguously determined by the comparison of experimental and theoretical CD spectra, which was affirmed by the X-ray crystal structural analysis of enantiomerically pure sulfonamide derivative CZ2.
@article{tani2020combined, title = {Combined Experimental and Theoretical Studies on Planar Chirality of Partially Overlapped C2-Symmetric [3.3](3,9)Dicarbazolophanes}, author = {Tani, Keita and Imafuku, Kanae and Masaki, Miyuki Eiraku and Kato, Haruka and Hori, Kazushige and Kubono, Koji and Taneda, Masatsugu and Harada, Takunori and Goto, Kenta and Tani, Fumito and Mori, Tadashi}, journal = {J. Phys. Chem. A}, volume = {124}, issue = {10}, pages = {2057--2063}, year = {2020}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.0c00286}, url = {https://doi.org/10.1021/acs.jpca.0c00286}, dimensions = {true}, tab = {paper}, } -
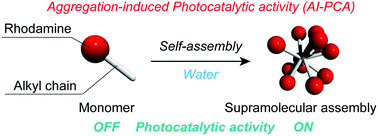 Aggregation-induced photocatalytic activity and efficient photocatalytic hydrogen evolution of amphiphilic rhodamines in waterHajime Shigemitsu*, Youhei Tani, Tomoe Tamemoto, Tadashi Mori, Xinxi Li, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki Kida*Chem. Sci., 2020, 11, 11843–11848.
Aggregation-induced photocatalytic activity and efficient photocatalytic hydrogen evolution of amphiphilic rhodamines in waterHajime Shigemitsu*, Youhei Tani, Tomoe Tamemoto, Tadashi Mori, Xinxi Li, Yasuko Osakada, Mamoru Fujitsuka, and Toshiyuki Kida*Chem. Sci., 2020, 11, 11843–11848.The development of photocatalysts is an essential task for clean energy generation and establishing a sustainable society. This paper describes the aggregation-induced photocatalytic activity (AI-PCA) of amphiphilic rhodamines and photocatalytic functions of the supramolecular assemblies. The supramolecular assemblies consisting of amphiphilic rhodamines with octadecyl alkyl chains exhibited significant photocatalytic activity under visible light irradiation in water, while the corresponding monomeric rhodamines did not exhibit photocatalytic activity. The studies on the photocatalytic mechanism by spectroscopic and microscopic analyses clearly demonstrated the AI-PCA of the rhodamines. Moreover, the supramolecular assemblies of the rhodamines exhibited excellent photocatalytic hydrogen evolution rates (up to 5.9 mmol g−1 h−1).
@article{shigemitsu2020aggregation, title = {Aggregation-induced photocatalytic activity and efficient photocatalytic hydrogen evolution of amphiphilic rhodamines in water}, author = {Shigemitsu, Hajime and Tani, Youhei and Tamemoto, Tomoe and Mori, Tadashi and Li, Xinxi and Osakada, Yasuko and Fujitsuka, Mamoru and Kida, Toshiyuki}, journal = {Chem. Sci.}, volume = {11}, issue = {43}, pages = {11843--11848}, year = {2020}, publisher = {Royal Society of Chemistry}, doi = {10.1039/d0sc04285d}, url = {https://doi.org/10.1039/d0sc04285d}, dimensions = {true}, tab = {paper}, } -
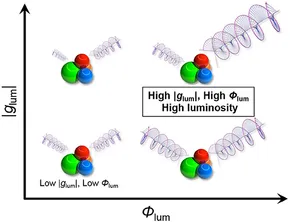 Irreverent nature of dissymmetry factor and quantum yield in circularly polarized luminescence of small organic moleculesYuya Nagata and Tadashi Mori*Front. Chem., 2020, 8, 448.
Irreverent nature of dissymmetry factor and quantum yield in circularly polarized luminescence of small organic moleculesYuya Nagata and Tadashi Mori*Front. Chem., 2020, 8, 448.Recently, a rational modification of small organic molecules has attracted considerable attention for designing advanced materials with enhanced circularly polarized luminescence (CPL) activity. A particular emphasis has been placed on fully allowed π-π* transition of rigid aromatic systems, due to their relatively superior emission properties or quantum yields of luminescence (Φlum). However, their dissymmetry factors (glum), differential left and right CPL intensities, are typically disappointingly low at least in one to two orders of magnitude. Truly useful organic CPL materials, rated by a circular polarization luminosity index (ΛCPL) per single molecule, possess both |glum| and Φlum values high. However, how to improve these two factors simultaneously with a proper molecular design is an open question. Here, we addressed this issue by theoretical and statistical inspection on a possible relation of the glum and Φlum values. According to the analysis, we propose simple, unpretentious, yet pertinent guidelines for designing superior organic CPL materials for the future with large ΛCPL values.
@article{nagata2020irreverent, title = {Irreverent nature of dissymmetry factor and quantum yield in circularly polarized luminescence of small organic molecules}, author = {Nagata, Yuya and Mori, Tadashi}, journal = {Front. Chem.}, volume = {8}, pages = {448}, year = {2020}, publisher = {Frontiers Media SA}, doi = {10.3389/fchem.2020.00448}, url = {https://doi.org/10.3389/fchem.2020.00448}, dimensions = {true}, tab = {review}, }
2019
-
 Sign control of circularly polarized luminescence based on geometric arrangement of fluorescent pyrene units in a binaphthyl scaffoldDaiki Kaji, Shintaro Ikeda, Kenya Takamura, Nobuo Tajima, Motohiro Shizuma, Tadashi Mori, Makoto Miyasaka*, and Yoshitane Imai*Chem. Lett., 2019, 48, 874–876.
Sign control of circularly polarized luminescence based on geometric arrangement of fluorescent pyrene units in a binaphthyl scaffoldDaiki Kaji, Shintaro Ikeda, Kenya Takamura, Nobuo Tajima, Motohiro Shizuma, Tadashi Mori, Makoto Miyasaka*, and Yoshitane Imai*Chem. Lett., 2019, 48, 874–876.The signs of circularly polarized luminescence and circular dichroism of binaphthyl-pyrene fluorophores with the same axial chirality can be controlled by altering the bonding positions of the two fluorescent pyrene units in the solution state. The relative geometrical arrangement of the two pyrene rings plays a substantial role in determining the signs of the observed chiroptical properties.
@article{kaji2019sign, title = {Sign control of circularly polarized luminescence based on geometric arrangement of fluorescent pyrene units in a binaphthyl scaffold}, author = {Kaji, Daiki and Ikeda, Shintaro and Takamura, Kenya and Tajima, Nobuo and Shizuma, Motohiro and Mori, Tadashi and Miyasaka, Makoto and Imai, Yoshitane}, journal = {Chem. Lett.}, volume = {48}, issue = {8}, pages = {874--876}, year = {2019}, publisher = {Oxford University Press}, doi = {10.1246/cl.190246}, url = {https://doi.org/10.1246/cl.190246}, dimensions = {true}, tab = {paper}, } -
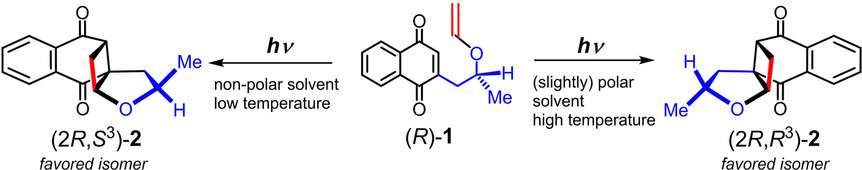 Diastereoselective Photocycloaddition Reaction of Vinyl Ether Tethered to 1, 4-NaphthoquinoneHiroki Ishikawa, Tim S Chung, Gaku Fukuhara, Hajime Shigemitsu, Toshiyuki Kida, Thorsten Bach, and Tadashi Mori*ChemPhotoChem, 2019, 3, 243–250.
Diastereoselective Photocycloaddition Reaction of Vinyl Ether Tethered to 1, 4-NaphthoquinoneHiroki Ishikawa, Tim S Chung, Gaku Fukuhara, Hajime Shigemitsu, Toshiyuki Kida, Thorsten Bach, and Tadashi Mori*ChemPhotoChem, 2019, 3, 243–250.The intramolecular asymmetric photocycloaddition between 1,4-naphthoquinone and vinyl ether with a small (R)-point-chiral group on the tether was studied. Under photoirradiation, [2+2] cyclobutane products were exclusively obtained for this intramolecular system. The effect of solvent polarity on the stereoselectivity was significant, with predominant formation of the (2R,S3) over the (2R,R3) isomer in non-polar solvents being reversed in slightly polar dichloromethane. The detailed temperature-dependent study revealed the dominant diastereo-differentiating processes, which were switched between the ground-state equilibrium, the relative rate of bond formation in the triplet manifold, as well as deactivation processes between the pro-(S3) and pro-(R3) precursors, depending on the temperature domain examined. The enthalpic contribution (ΔΔH≠) was always compensated by the entropic factor (ΔΔS≠), implying the importance of solvation on the diastereo-differentiation steps. The mechanism of photocycloaddition, especially for the face-selective processes, is thoroughly discussed, which is supported by quantum chemical calculations on the ground-state circular dichroism (CD) spectral behavior as well as on the diastereomeric transition states in the triplet excited state.
@article{ishikawa2019diastereoselective, title = {Diastereoselective Photocycloaddition Reaction of Vinyl Ether Tethered to 1, 4-Naphthoquinone}, author = {Ishikawa, Hiroki and Chung, Tim S and Fukuhara, Gaku and Shigemitsu, Hajime and Kida, Toshiyuki and Bach, Thorsten and Mori, Tadashi}, journal = {ChemPhotoChem}, volume = {3}, issue = {5}, pages = {243--250}, year = {2019}, publisher = {Wiley Online Library}, doi = {10.1002/cptc.201900022}, url = {https://doi.org/10.1002/cptc.201900022}, dimensions = {true}, tab = {paper}, } -
 Transient circular dichroism measurement of the excited triplet state of pristine hexahelicene in solution at room temperatureMakoto Kuronuma, Takehito Sato, Yasuyuki Araki*, Tadashi Mori, Seiji Sakamoto, Yoshihisa Inoue, Osamu Ito, and Takehiko Wada*Chem. Lett., 2019, 48, 357–360.
Transient circular dichroism measurement of the excited triplet state of pristine hexahelicene in solution at room temperatureMakoto Kuronuma, Takehito Sato, Yasuyuki Araki*, Tadashi Mori, Seiji Sakamoto, Yoshihisa Inoue, Osamu Ito, and Takehiko Wada*Chem. Lett., 2019, 48, 357–360.The intramolecular asymmetric photocycloaddition between 1,4-naphthoquinone and vinyl ether with a small (R)-point-chiral group on the tether was studied. Under photoirradiation, [2+2] cyclobutane products were exclusively obtained for this intramolecular system. The effect of solvent polarity on the stereoselectivity was significant, with predominant formation of the (2R,S3) over the (2R,R3) isomer in non-polar solvents being reversed in slightly polar dichloromethane. The detailed temperature-dependent study revealed the dominant diastereo-differentiating processes, which were switched between the ground-state equilibrium, the relative rate of bond formation in the triplet manifold, as well as deactivation processes between the pro-(S3) and pro-(R3) precursors, depending on the temperature domain examined. The enthalpic contribution (ΔΔH≠) was always compensated by the entropic factor (ΔΔS≠), implying the importance of solvation on the diastereo-differentiation steps. The mechanism of photocycloaddition, especially for the face-selective processes, is thoroughly discussed, which is supported by quantum chemical calculations on the ground-state circular dichroism (CD) spectral behavior as well as on the diastereomeric transition states in the triplet excited state.
@article{kuronuma2019transient, title = {Transient circular dichroism measurement of the excited triplet state of pristine hexahelicene in solution at room temperature}, author = {Kuronuma, Makoto and Sato, Takehito and Araki, Yasuyuki and Mori, Tadashi and Sakamoto, Seiji and Inoue, Yoshihisa and Ito, Osamu and Wada, Takehiko}, journal = {Chem. Lett.}, volume = {48}, issue = {4}, pages = {357--360}, year = {2019}, publisher = {Oxford University Press}, doi = {10.1246/cl.190012}, url = {https://doi.org/10.1246/cl.190012}, dimensions = {true}, tab = {paper}, } -
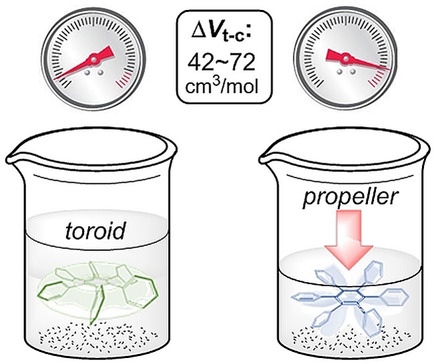 Hydrostatic pressure on toroidal interaction and propeller chirality of hexaarylbenzenes: explicit solvent effects on differential volumes in methylcyclohexane and hexaneTomoyo Kosaka, Satono Iwai, Gaku Fukuhara*, Yoshitane Imai, and Tadashi Mori*Chem. Eur. J., 2019, 25, 2011–2018.
Hydrostatic pressure on toroidal interaction and propeller chirality of hexaarylbenzenes: explicit solvent effects on differential volumes in methylcyclohexane and hexaneTomoyo Kosaka, Satono Iwai, Gaku Fukuhara*, Yoshitane Imai, and Tadashi Mori*Chem. Eur. J., 2019, 25, 2011–2018.A unique and effective interaction between the peripheral aromatic blades makes hexaarylbenzenes (HABs) attractive in fundamental research as well as for various applications such as molecular wires, sensors, and supramolecular assemblies. The chiroptical responses of HABs are susceptible to environmental factors such as solvent and temperature owing to the dynamic conformational transitions between the conformers. In this study, pressure dependence on the propeller chiral HABs in two different solvents was studied in detail. The effective differential volumes for two different equilibria were determined by quantitative analyses of CD spectra, affording very large differential volumes from the propeller to toroidal conformer (ΔVT-C) of +43 and +42 cm3 mol−1, for H2 and H6, respectively, in methylcyclohexane. The value of H6 was further enhanced to +72 cm3 mol−1 in hexane, the largest value for the typical unimolecular conformational change. Such a response of propeller chirality in HABs is expedient in designing more advanced piezo-sensitive materials.
@article{kosaka2019hydrostatic, title = {Hydrostatic pressure on toroidal interaction and propeller chirality of hexaarylbenzenes: explicit solvent effects on differential volumes in methylcyclohexane and hexane}, author = {Kosaka, Tomoyo and Iwai, Satono and Fukuhara, Gaku and Imai, Yoshitane and Mori, Tadashi}, journal = {Chem. Eur. J.}, volume = {25}, issue = {8}, pages = {2011--2018}, year = {2019}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201804688}, url = {https://doi.org/10.1002/chem.201804688}, dimensions = {true}, tab = {paper}, } -
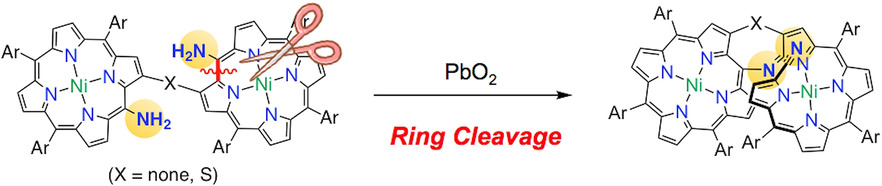 Selective Formation of Helical Tetrapyrrin-Fused Porphyrins by Oxidation of β-to-β Linked meso-Aminoporphyrin DimersKeisuke Fujimoto, Daiki Shimizu, Tadashi Mori, Yuanyuan Li, Mingbo Zhou, Jianxin Song, and Atsuhiro Osuka*Chem. Eur. J., 2019, 25, 1711–1715.
Selective Formation of Helical Tetrapyrrin-Fused Porphyrins by Oxidation of β-to-β Linked meso-Aminoporphyrin DimersKeisuke Fujimoto, Daiki Shimizu, Tadashi Mori, Yuanyuan Li, Mingbo Zhou, Jianxin Song, and Atsuhiro Osuka*Chem. Eur. J., 2019, 25, 1711–1715.Oxidation of β-to-β directly linked and sulfur-bridged meso-amino NiII-porphyrin dimers with PbO2 gave helical tetrapyrrin (biliverdin analogue)-fused NiII-porphyrins. These ring cleaving reactions differ markedly from the previously reported oxidation of a β–β linked NiII-porphyrin dimer carrying one amino group, which gave an azepine-fused porphyrin dimer. The tetrapyrrin-fused NiII-porphyrins display intense NIR absorption bands at 1200–1400 nm and reversible redox processes because of the highly π-conjugated networks and rigid structures. These tetrapyrrin-fused NiII-porphyrins were separated to stable enantiomers, which showed clear Cotton effects in their CD spectra with Δϵ of 102 order.
@article{fujimoto2019selective, title = {Selective Formation of Helical Tetrapyrrin-Fused Porphyrins by Oxidation of β-to-β Linked meso-Aminoporphyrin Dimers}, author = {Fujimoto, Keisuke and Shimizu, Daiki and Mori, Tadashi and Li, Yuanyuan and Zhou, Mingbo and Song, Jianxin and Osuka, Atsuhiro}, journal = {Chem. Eur. J.}, volume = {25}, issue = {7}, pages = {1711--1715}, year = {2019}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201805659}, url = {https://doi.org/10.1002/chem.201805659}, dimensions = {true}, tab = {paper}, } -
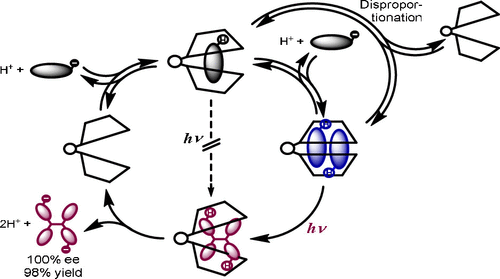 An ultimate stereocontrol in supramolecular photochirogenesis: photocyclodimerization of 2-anthracenecarboxylate mediated by sulfur-linked β-cyclodextrin dimersJiecheng Ji, Wanhua Wu, Wenting Liang, Guo Cheng, Ryohei Matsushita, Zhiqiang Yan, Xueqin Wei, Ming Rao, De-Qi Yuan, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cheng Yang*J. Am. Chem. Soc., 2019, 141, 9225–9238.
An ultimate stereocontrol in supramolecular photochirogenesis: photocyclodimerization of 2-anthracenecarboxylate mediated by sulfur-linked β-cyclodextrin dimersJiecheng Ji, Wanhua Wu, Wenting Liang, Guo Cheng, Ryohei Matsushita, Zhiqiang Yan, Xueqin Wei, Ming Rao, De-Qi Yuan, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cheng Yang*J. Am. Chem. Soc., 2019, 141, 9225–9238.Stereoisomeric β-cyclodextrin (CD) dimers linked with a sulfur atom or an arene spacer were designed to create a tethered dual CD capsule for precisely manipulating the regio- and enantioselectivities of the photocyclodimerization of 2-anthracenecarboxylate (AC) to four stereoisomeric classical 9,10:9′,10′-cyclodimers and two nonclassical 5,8:9′,10′-cyclodimers. Among the dimeric CD hosts prepared, exo-3-thia-β-CD dimer formed 1:1 and 1:2 host–guest complexes with AC in aqueous solutions, the former of which hindered but the latter facilitated the AC photocyclodimerization with regio- and enantioselectivities much higher than those obtained with native β-CD or the rest of the β-CD dimers. The stereochemical outcomes turned out to be highly sensitive to and hence critically manipulable by the linking position and configuration of the connected saccharide units and the linker length, as well as the external variants, such as temperature, pH, and added salt. Eventually, the photocyclodimerization of AC mediated by the dimeric β-CD host gave enantiopure syn-head-to-tail-9,10:9′,10′-cyclodimer in 97–98% yield in a pH 5.1 buffer solution at 0.5 °C and also in an aqueous CsCl solution at −20 °C.
@article{ji2019ultimate, title = {An ultimate stereocontrol in supramolecular photochirogenesis: photocyclodimerization of 2-anthracenecarboxylate mediated by sulfur-linked β-cyclodextrin dimers}, author = {Ji, Jiecheng and Wu, Wanhua and Liang, Wenting and Cheng, Guo and Matsushita, Ryohei and Yan, Zhiqiang and Wei, Xueqin and Rao, Ming and Yuan, De-Qi and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa and Yang, Cheng}, journal = {J. Am. Chem. Soc.}, volume = {141}, number = {23}, pages = {9225--9238}, year = {2019}, publisher = {ACS Publications}, doi = {10.1021/jacs.9b01993}, url = {https://doi.org/10.1021/jacs.9b01993}, dimensions = {true}, tab = {paper}, }
2018
-
 Spiroborate-based double-stranded helicates: meso-to-racemo isomerization and ion-triggered springlike motion of the racemo-helicateNaoki Ousaka, Kaori Shimizu, Yoshimasa Suzuki, Takuya Iwata, Manabu Itakura, Daisuke Taura, Hiroki Iida, Yoshio Furusho, Tadashi Mori, and Eiji Yashima*J. Am. Chem. Soc., 2018, 140, 17027–17039.
Spiroborate-based double-stranded helicates: meso-to-racemo isomerization and ion-triggered springlike motion of the racemo-helicateNaoki Ousaka, Kaori Shimizu, Yoshimasa Suzuki, Takuya Iwata, Manabu Itakura, Daisuke Taura, Hiroki Iida, Yoshio Furusho, Tadashi Mori, and Eiji Yashima*J. Am. Chem. Soc., 2018, 140, 17027–17039.A one-handed double-stranded spiroborate helicate exhibits a unique reversible extension–contraction motion coupled with a twisting motion in one direction triggered by binding and release of a Na+ ion while retaining its handedness. Here we report that an extended meso-helicate was also produced together with the racemo-helicate, and the meso-helicate was readily converted to the racemo-helicate assisted by a Na+ ion as a template in the presence of water. The thermodynamic analyses of the ion-triggered springlike motion of the racemo-helicate using a series of monovalent cations with different sizes revealed that the association constants of the extended racemo-helicate decreased in the following order: Li+ > Na+ > NH4+ > Ag+ ≥ K+ > Cs+ > Rb+, which roughly agrees with the reverse order of their ionic radii except for the NH4+ ion due to the more elongated contracted helicates when complexed with larger cations as supported by the crystal and DFT calculated structures. The one-handed contracted helicates showed characteristic CD spectra depending on the cation species due to the differences in their contracted helical structures, and its absolute handedness of the spiroborate helicate was determined by X-ray crystallography. The kinetic studies of the springlike motions of the racemo-helicate showed that the exchange rate between the extended and contracted helicates tend to increase in the following order: Li+ < Na+ < K+ ≤ NH4+ < Rb+ < Cs+ < Ag+ as anticipated from the association constants, being in good agreement with the order of the cation sizes except for Ag+.
@article{ousaka2018spiroborate, title = {Spiroborate-based double-stranded helicates: meso-to-racemo isomerization and ion-triggered springlike motion of the racemo-helicate}, author = {Ousaka, Naoki and Shimizu, Kaori and Suzuki, Yoshimasa and Iwata, Takuya and Itakura, Manabu and Taura, Daisuke and Iida, Hiroki and Furusho, Yoshio and Mori, Tadashi and Yashima, Eiji}, journal = {J. Am. Chem. Soc.}, volume = {140}, issue = {49}, pages = {17027--17039}, year = {2018}, publisher = {ACS Publications}, doi = {10.1021/jacs.8b08268}, url = {https://doi.org/10.1021/jacs.8b08268}, dimensions = {true}, tab = {paper}, } -
 Symmetry-based rational design for boosting chiroptical responsesHiroki Tanaka, Mina Ikenosako, Yuka Kato, Michiya Fujiki, Yoshihisa Inoue, and Tadashi Mori*Communications Chemistry, 2018, 1, 38.
Symmetry-based rational design for boosting chiroptical responsesHiroki Tanaka, Mina Ikenosako, Yuka Kato, Michiya Fujiki, Yoshihisa Inoue, and Tadashi Mori*Communications Chemistry, 2018, 1, 38.Chiral molecules play indispensable roles in advanced materials and technologies. Nevertheless, no conventional, yet reliable logical strategies are available for designing chiral molecules of desired chiroptical properties. Here, we propose a general protocol for rationally aligning multiple chiral units to boost the chiroptical responses, using hexahelicene as a prototype. In this proof-of-concept study, we align two hexahelicenes in various orientations and examine by theoretical calculations to predict the best chiroptical performance for X-shaped and S-shaped double hexahelicenes. We synthesize and optically resolve both double hexahelicenes and show that they exhibit more than a twofold increase in intensity of circular dichroism and circularly polarized luminescence, experimentally validating the protocol. The enhanced chiroptical responses are theoretically assignable to the electric and magnetic transition dipole moments of component hexahelicenes aligned in the correct symmetry. A guiding principle for designing advanced molecular and supramolecular chiral materials is further discussed.
@article{tanaka2018symmetry, title = {Symmetry-based rational design for boosting chiroptical responses}, author = {Tanaka, Hiroki and Ikenosako, Mina and Kato, Yuka and Fujiki, Michiya and Inoue, Yoshihisa and Mori, Tadashi}, journal = {Communications Chemistry}, volume = {1}, issue = {1}, pages = {38}, year = {2018}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s42004-018-0035-x}, url = {https://doi.org/10.1038/s42004-018-0035-x}, dimensions = {true}, tab = {paper}, } -
 Significant Enhancement of Absorption and Luminescence Dissymmetry Factors in the Far-Red Region: A Zinc (II) Homoleptic Helicate Formed by a Pair of Achiral Dipyrromethene LigandsHiroaki Ito, Hayato Sakai, Yoshinori Okayasu, Junpei Yuasa, Tadashi Mori*, and Taku Hasobe*Chem. Eur. J., 2018, 24, 16889–16894.
Significant Enhancement of Absorption and Luminescence Dissymmetry Factors in the Far-Red Region: A Zinc (II) Homoleptic Helicate Formed by a Pair of Achiral Dipyrromethene LigandsHiroaki Ito, Hayato Sakai, Yoshinori Okayasu, Junpei Yuasa, Tadashi Mori*, and Taku Hasobe*Chem. Eur. J., 2018, 24, 16889–16894.A homoleptic zinc(II) helicate organized by a pair of achiral dipyrromethene ligands through zinc(II) coordination was synthesized to evaluate the chiroptical properties. This zinc complex showed strong exciton-coupled chiroptical responses from the helical configuration with a large absorption dissymmetry factor |gabs| (up to 0.20). More importantly, intense polarized luminescence in the far-red region (700–850 nm) with a fluorescence quantum yield ΦFL of 0.23 was observed for this helicate with a dissymmetry factor |glum| of 0.022, the largest value among rare-earth- and precious-metal-free small molecules. These unprecedentedly large g values were supported by theoretical calculations.
@article{ito2018significant, title = {Significant Enhancement of Absorption and Luminescence Dissymmetry Factors in the Far-Red Region: A Zinc (II) Homoleptic Helicate Formed by a Pair of Achiral Dipyrromethene Ligands}, author = {Ito, Hiroaki and Sakai, Hayato and Okayasu, Yoshinori and Yuasa, Junpei and Mori, Tadashi and Hasobe, Taku}, journal = {Chem. Eur. J.}, volume = {24}, issue = {63}, pages = {16889--16894}, year = {2018}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201804171}, url = {https://doi.org/10.1002/chem.201804171}, dimensions = {true}, tab = {paper}, } -
 Combined Experimental and Theoretical Study on Circular Dichroism and Circularly Polarized Luminescence of Configurationally Robust D3-Symmetric Triple PentaheliceneHiroki Tanaka, Yuka Kato, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2018, 122, 7378–7384.
Combined Experimental and Theoretical Study on Circular Dichroism and Circularly Polarized Luminescence of Configurationally Robust D3-Symmetric Triple PentaheliceneHiroki Tanaka, Yuka Kato, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2018, 122, 7378–7384.Pentahelicene (PH) exhibits the largest absorption (gabs) and luminescence (glum) dissymmetry factors among the helicene family but is configurationally and (photo)chemically labile, encumbering its application to chiroptical materials. To bypass the pitfalls, three PH units are merged in a single molecule to build D3-symmetric triple pentahelicene, hexabenzotriphenylene (HBT), which attains indeed the configurational and (photo)chemical robustness through equilibrium with a C2-symmetric conformer that interrupts the racemization and photocyclization. UV–vis, circular dichroism (CD), and circularly polarized luminescence (CPL) spectral examinations reveal the significantly larger gabs and glum values for HBT than for any of configurationally robust single [n]helicene (n ≥ 6) and C2-symmetric triple pentahelicene, trinaphthotriphenylene (TNT). Theoretical calculations precisely reproduce the main features of the experimental CD and CPL spectra of PH, HBT, and TNT, and the relevant electric and magnetic transition moments and their mutual angles well rationalize the relative CD and CPL intensities of all the single and triple pentahelicenes.
@article{tanaka2018combined, title = {Combined Experimental and Theoretical Study on Circular Dichroism and Circularly Polarized Luminescence of Configurationally Robust D3-Symmetric Triple Pentahelicene}, author = {Tanaka, Hiroki and Kato, Yuka and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Phys. Chem. A}, volume = {122}, issue = {37}, pages = {7378--7384}, year = {2018}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.8b05247}, url = {https://doi.org/10.1021/acs.jpca.8b05247}, dimensions = {true}, tab = {paper}, } -
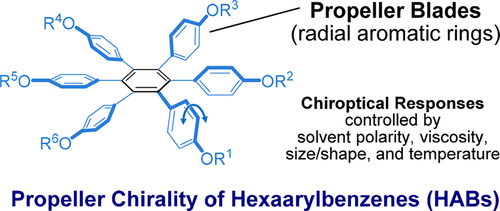 Solvent and temperature effects on dynamics and chiroptical properties of propeller chirality and toroidal interaction of hexaarylbenzenesTomoyo Kosaka, Satono Iwai, Yoshihisa Inoue, Toshiyuki Moriuchi, and Tadashi Mori*The Journal of Physical Chemistry A, 2018, 122, 7455–7463.
Solvent and temperature effects on dynamics and chiroptical properties of propeller chirality and toroidal interaction of hexaarylbenzenesTomoyo Kosaka, Satono Iwai, Yoshihisa Inoue, Toshiyuki Moriuchi, and Tadashi Mori*The Journal of Physical Chemistry A, 2018, 122, 7455–7463.Because of the unique interaction of radial aromatic blades, propeller-shaped hexaarylbenzenes (HABs) attract much research interest and find various practical applications. By introducing a small point-chiral group at the tip of aromatic blade(s), HAB becomes propeller-chiral to exhibit strong Cotton effects. Because of the dynamic nature of propeller chirality, the chiroptical properties of HAB critically responded to minute changes in the environment. Using a series of chiral HABs with one to six (R)-1-methylpropyloxy substituent(s) introduced at the blade tip, we elucidated how the smallest chiral auxiliary at the HAB periphery progressively and cooperatively boosts the overall chiroptical properties and also how subtle changes in temperature and solvent structure affect the propeller dynamics and thus the chiroptical responses. The unique features of propeller-chiral HABs further enabled us to switch on/off their circularly polarized luminescence.
@article{kosaka2018solvent, title = {Solvent and temperature effects on dynamics and chiroptical properties of propeller chirality and toroidal interaction of hexaarylbenzenes}, author = {Kosaka, Tomoyo and Iwai, Satono and Inoue, Yoshihisa and Moriuchi, Toshiyuki and Mori, Tadashi}, journal = {The Journal of Physical Chemistry A}, volume = {122}, issue = {37}, pages = {7455--7463}, year = {2018}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.8b06535}, url = {https://doi.org/10.1021/acs.jpca.8b06535}, dimensions = {true}, tab = {paper}, } -
 Synthesis, structures, and optical properties of azahelicene derivatives and unexpected formation of azahepta [8] circulenesFengkun Chen, Takayuki Tanaka*, Tadashi Mori, and Atsuhiro Osuka*Chem. Eur. J., 2018, 24, 7489–7497.
Synthesis, structures, and optical properties of azahelicene derivatives and unexpected formation of azahepta [8] circulenesFengkun Chen, Takayuki Tanaka*, Tadashi Mori, and Atsuhiro Osuka*Chem. Eur. J., 2018, 24, 7489–7497.Polycyclic heteroaromatic compounds including pyrrole units are promising functional scaffolds owing to their electron-rich nature, bright fluorescence, and applicability to anion recognition at the pyrrolic hydrogen atom. We report herein the effective synthesis of pseudo-aza[5]helicene and aza[7]helicene derivatives, and unexpected formation of azahepta[8]circulenes by oxidative fusion reactions. By choosing reaction conditions and peripheral substituents attached at the terminal indole moieties, we obtained aza[7]helicenes 10 a–c and azahepta[8]circulenes 11 a,c selectively in moderate to good yields. Their solid-state structures have been revealed by X-ray diffraction analysis. UV/Vis absorption, emission, and cyclic voltammetry of these compounds were studied in comparison with those of previously reported tetraaza[8]circulene (TA8C), a symmetric and planar molecule. Furthermore, the enantiomeric separation of dimethyl-substituted aza[7]helicene 10 b was achieved, and the racemization kinetics have been elucidated both theoretically and experimentally. This work illustrates a wealth of advantages of pyrrole incorporation into the polycyclic aromatic scaffolds in terms of synthetic aspects, structural variation, and optical tuning.
@article{chen2018synthesis, title = {Synthesis, structures, and optical properties of azahelicene derivatives and unexpected formation of azahepta [8] circulenes}, author = {Chen, Fengkun and Tanaka, Takayuki and Mori, Tadashi and Osuka, Atsuhiro}, journal = {Chem. Eur. J.}, volume = {24}, issue = {29}, pages = {7489--7497}, year = {2018}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201800617}, url = {https://doi.org/10.1002/chem.201800617}, dimensions = {true}, tab = {paper}, } -
 1, 8-Diphenyl-9, 10-Bis (arylethynyl) phenanthrenes: Synthesis, Distorted Structure, and Optical PropertiesAkihito Konishi*, Atsushi Morinaga, Gaku Fukuhara, Masaki Nishijima, Tadashi Mori, Toshiyuki Kida, and Makoto Yasuda*Chem. Eur. J., 2018, 24, 6625–6631.
1, 8-Diphenyl-9, 10-Bis (arylethynyl) phenanthrenes: Synthesis, Distorted Structure, and Optical PropertiesAkihito Konishi*, Atsushi Morinaga, Gaku Fukuhara, Masaki Nishijima, Tadashi Mori, Toshiyuki Kida, and Makoto Yasuda*Chem. Eur. J., 2018, 24, 6625–6631.The synthesis and optical properties of 1,8-diphenyl-9,10-bis(arylethynyl)phenanthrenes, which are distorted phenanthrenes, are reported. The presence of the two phenyl groups at the 1,8-positions of phenanthrene significantly distorts the molecular geometries, as was evidenced by X-ray crystallography. The congested substitution pattern in the K region results in a distorted aromatic framework, which leads to a redshift in the emission spectrum. These observations are in stark contrast to 9,10-bis(phenylethynyl)phenanthrene with no phenyl groups at the 1,8-positions. A large Stokes shift suggested extensive structural relaxation between the phenyl and arylethynyl units in the excited state, which was supported by theoretical calculations.
@article{konishi20181, title = {1, 8-Diphenyl-9, 10-Bis (arylethynyl) phenanthrenes: Synthesis, Distorted Structure, and Optical Properties}, author = {Konishi, Akihito and Morinaga, Atsushi and Fukuhara, Gaku and Nishijima, Masaki and Mori, Tadashi and Kida, Toshiyuki and Yasuda, Makoto}, journal = {Chem. Eur. J.}, volume = {24}, issue = {25}, pages = {6625--6631}, year = {2018}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201800617}, url = {https://doi.org/10.1002/chem.201800617}, dimensions = {true}, tab = {paper}, } -
 Circularly polarized luminescence and circular dichroisms in small organic molecules: correlation between excitation and emission dissymmetry factorsHiroki Tanaka, Yoshihisa Inoue, and Tadashi Mori*ChemPhotoChem, 2018, 2, 386–402.
Circularly polarized luminescence and circular dichroisms in small organic molecules: correlation between excitation and emission dissymmetry factorsHiroki Tanaka, Yoshihisa Inoue, and Tadashi Mori*ChemPhotoChem, 2018, 2, 386–402.Prompted by the recent rapid growth of interest in circularly polarized luminescence (CPL) of organic molecules, we have collected all the reliable CPL, as well as the corresponding circular dichroism (CD), data measured in fluid solutions. To analyze the correlation between CPL and CD, we employed the absorption and luminescence dissymmetry factors (gabs and glum) of the π–π* transition reported for chiral organic molecules of various categories, including planar chiral cyclophanes and helicenes, axially chiral biaryls and spiro compounds, and point- and axially chiral BODIPY derivatives. In rigid π-systems, the absorption and fluorescence spectra are often mirror images of each other with a small Stokes shift, reflecting the minimal conformational relaxation in the emissive excited state, which should also affect the chiroptical properties in the excited state and be better sensed by CPL. However, no comprehensive efforts have hitherto been made to correlate the two relevant chiroptical properties, i.e. CPL versus CD, and also to quantitatively elucidate the effects of conformational relaxation in the excited state on the CPL behavior. The global linear regression analysis of all the reported gabs and glum values, though fairly scattered (see TOC), led us to a quantitative relationship: |glum|=0.81×|gabs| (r2=0.60), which demonstrates that the CPL dissymmetry factor is proportional to, and smaller than, the CD dissymmetry factor. A closer look revealed that the slope of the plot, or the proportional coefficient, is a critical function of the class of compounds, varying from 0.99 for cyclophanes to 0.93 for biaryls, to 0.77 for BODIPYs, and then to 0.61 for helicenes/helicenoids. The scattered glum–gabs plot and the general trend glum≤gabs appear to be inherent to the CPL of organic molecules in their isolated states, originating from the conformational flexibility, vibrational contribution, and Stokes shift that differ in each category.
@article{tanaka2018circularly, title = {Circularly polarized luminescence and circular dichroisms in small organic molecules: correlation between excitation and emission dissymmetry factors}, author = {Tanaka, Hiroki and Inoue, Yoshihisa and Mori, Tadashi}, journal = {ChemPhotoChem}, volume = {2}, issue = {5}, pages = {386--402}, year = {2018}, publisher = {Wiley Online Library}, doi = {10.1002/cptc.201800015}, url = {https://doi.org/10.1002/cptc.201800015}, dimensions = {true}, tab = {review}, } -
 Entropy-Driven Diastereoselectivity Improvement in the Paternò–Büchi Reaction of 1-Naphthyl Aryl Ethenes with a Chiral Cyanobenzoate through Remote AlkylationKeisuke Nagasaki, Yoshihisa Inoue, and Tadashi Mori*Angew. Chem. Int. Ed., 2018, 57, 4880–4885.
Entropy-Driven Diastereoselectivity Improvement in the Paternò–Büchi Reaction of 1-Naphthyl Aryl Ethenes with a Chiral Cyanobenzoate through Remote AlkylationKeisuke Nagasaki, Yoshihisa Inoue, and Tadashi Mori*Angew. Chem. Int. Ed., 2018, 57, 4880–4885.The precise stereocontrol of photocycloaddition reactions is still a significant challenge owing to their mechanistic complexity and the involvement of highly reactive and short-lived intermediates. Attempts have hitherto been made through structural modifications, mostly by introducing steric conflicts, to increase the difference between the enthalpic barriers. Herein, we show that entropy plays a crucial role in influencing the diastereoselectivity of a Paternò–Büchi reaction. Remote meta alkylation of the donor caused nominal changes in its photophysical properties as well as those of the exciplexes derived thereof. Nevertheless, the diastereomeric excess of the oxetane product was greatly improved by about 40%. This enhancement, which is not accompanied by any significant changes in the photophysical properties, is difficult to rationalize by conventional enthalpic control concepts based on repulsive steric and/or attractive intermolecular interactions as well as electronic perturbations. Differential activation parameters and compensatory enthalpy–entropy relationships revealed that the diastereoselectivity enhancement is not simply enthalpic but also entropic in origin.
@article{nagasaki2018entropy, title = {Entropy-Driven Diastereoselectivity Improvement in the Paternò--Büchi Reaction of 1-Naphthyl Aryl Ethenes with a Chiral Cyanobenzoate through Remote Alkylation}, author = {Nagasaki, Keisuke and Inoue, Yoshihisa and Mori, Tadashi}, journal = {Angew. Chem. Int. Ed.}, volume = {57}, issue = {18}, pages = {4880--4885}, year = {2018}, publisher = {Wiley Online Library}, doi = {10.1002/anie.201801330}, url = {https://doi.org/10.1002/anie.201801330}, dimensions = {true}, tab = {paper}, } -
 Supramolecular photochirogenesis driven by higher-order complexation: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate to slipped cyclodimers via a 2: 2 complex with β-cyclodextrinXueqin Wei, Wanhua Wu, Ryohei Matsushita, Zhiqiang Yan, Dayang Zhou, Jason J Chruma, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cheng Yang*J. Am. Chem. Soc., 2018, 140, 3959–3974.
Supramolecular photochirogenesis driven by higher-order complexation: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate to slipped cyclodimers via a 2: 2 complex with β-cyclodextrinXueqin Wei, Wanhua Wu, Ryohei Matsushita, Zhiqiang Yan, Dayang Zhou, Jason J Chruma, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cheng Yang*J. Am. Chem. Soc., 2018, 140, 3959–3974.Chiral slipped 5,8:9′,10′-cyclodimers were preferentially produced over classical 9,10:9′,10′-cyclodimers upon supramolecular photocyclodimerization of 2-anthracenecarboxylate (AC) mediated by β-cyclodextrin (β-CD). This photochirogenic route to the slipped cyclodimers, exclusively head-to-tail (HT) and highly enantioselective, has long been overlooked in foregoing studies but is dominant in reality and is absolutely supramolecularly activated by 2:2 complexation of AC with β-CD. The intricate structural and photophysical aspects of this higher-order complexation-triggered process have been comprehensively elucidated, while the absolute configurations of the slipped cyclodimers have been unambiguously assigned by comparing the experimental and theoretical circular dichroism spectra. In the 2:2 complex, two ACs packed in a dual β-CD capsule are not fully overlapped with each other but are only partially stacked in a slipped anti- or syn-HT manner. Hence, they do not spontaneously cyclodimerize upon photoexcitation but instead emit long-lived excimer fluorescence at wavelengths slightly longer than the monomer fluorescence, indicating that the slipped excimer is neither extremely reactive nor completely relaxed in conformation and energy. Because of the slipped conformation of the AC pair in the soft capsule, the subsequent photocyclodimerization becomes manipulable by various internal or external factors, such as temperature, pressure, added salt, and host modification, enabling us to exclusively obtain the slipped cyclodimers with high regio- and enantioselectivities. In this supramolecularly driven photochirogenesis, the dual β-CD capsule functions as a chiral organophotocatalyst to trigger and accelerate the nonclassical photochirogenic route to slipped cyclodimers by preorganizing the conformation of the encapsulated AC pair, formally mimicking a catalytic antibody.
@article{wei2018supramolecular, title = {Supramolecular photochirogenesis driven by higher-order complexation: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate to slipped cyclodimers via a 2: 2 complex with β-cyclodextrin}, author = {Wei, Xueqin and Wu, Wanhua and Matsushita, Ryohei and Yan, Zhiqiang and Zhou, Dayang and Chruma, Jason J and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa and Yang, Cheng}, journal = {J. Am. Chem. Soc.}, volume = {140}, issue = {11}, pages = {3959--3974}, year = {2018}, publisher = {ACS Publications}, doi = {10.1021/jacs.7b12085}, url = {https://doi.org/10.1021/jacs.7b12085}, dimensions = {true}, tab = {paper}, } -
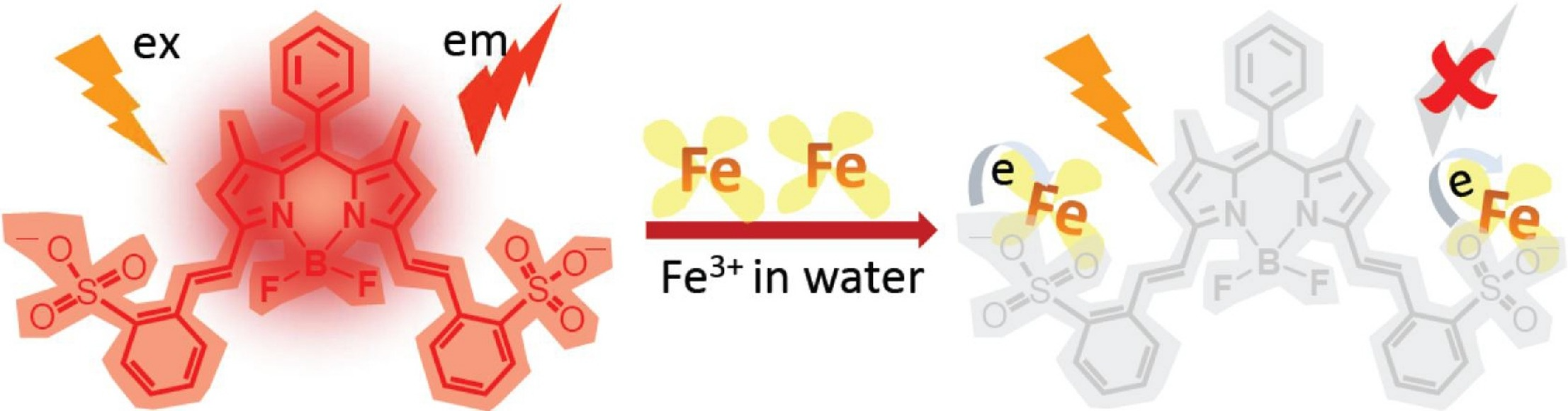 A BODIPY-based near infrared fluorescent probe for Fe3+ in waterJiecheng Ji, Syama Sundar Chereddy, Yi Ren, Xingming Chen, Dan Su, Zhihui Zhong, Tadashi Mori, Yoshihisa Inoue*, Wanhua Wu, and Cheng Yang*J. Photochem. Photobiol. A: Chem., 2018, 355, 78–83.
A BODIPY-based near infrared fluorescent probe for Fe3+ in waterJiecheng Ji, Syama Sundar Chereddy, Yi Ren, Xingming Chen, Dan Su, Zhihui Zhong, Tadashi Mori, Yoshihisa Inoue*, Wanhua Wu, and Cheng Yang*J. Photochem. Photobiol. A: Chem., 2018, 355, 78–83.For highly sensitive and selective detection of biologically important Fe3+ in aqueous media, a novel near-infrared (NIR) fluorescent probe (λem = 627 nm; ΦF = 0.47) based on boron-dipyrromethene (BODIPY) was developed. Employing a pair of 2-sulfonatostyryl sidearms attached to the BODIPY core as a reversible chelator specific to Fe3+, the sensor achieved a high selectivity toward Fe3+ over a wide range of metal ions, including Li+, Na+, K+, Mg2+, Ca2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Hg2+, Al3+, and Cr3+. Furthermore, the quenched NIR fluorescence was immediately recovered upon addition of sodium ascorbate as a reductant or EDTA as a stronger chelator.
@article{ji2018bodipy, title = {A BODIPY-based near infrared fluorescent probe for Fe3+ in water}, author = {Ji, Jiecheng and Chereddy, Syama Sundar and Ren, Yi and Chen, Xingming and Su, Dan and Zhong, Zhihui and Mori, Tadashi and Inoue, Yoshihisa and Wu, Wanhua and Yang, Cheng}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {355}, pages = {78--83}, year = {2018}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2017.10.043}, url = {https://doi.org/10.1016/j.jphotochem.2017.10.043}, dimensions = {true}, tab = {paper}, } -
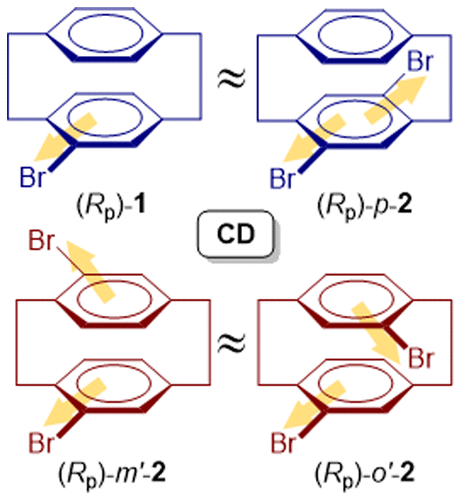 Circular Dichroisms of Mono-and Dibromo [2.2] paracyclophanes: A Combined Experimental and Theoretical StudyMitsunobu Toda, Yoshihisa Inoue, and Tadashi Mori*ACS Omega, 2018, 3, 22–29.
Circular Dichroisms of Mono-and Dibromo [2.2] paracyclophanes: A Combined Experimental and Theoretical StudyMitsunobu Toda, Yoshihisa Inoue, and Tadashi Mori*ACS Omega, 2018, 3, 22–29.Circular dichroisms (CDs) of planar chiral 4-bromo[2.2]paracyclophane (1) and three isomeric dibromo[2.2]paracyclophanes (p-2, m′-2, and o′-2) were investigated experimentally and theoretically. They all exhibited strong multisignate Cotton effects (CEs) at the 1Lb, 1La, and 1B transitions of the component (bromo)benzene chromophore and were comparable to each other. For all of the cyclophanes examined, the enantiomer that eluted earlier from a chiral high-performance liquid chromatography column (Chiralcel IA or IB) exhibited negative and positive CEs at the 1Lb and 1La bands, respectively, which were followed by a more complicated pattern of CDs at the higher-energy bands. These CD features were well reproduced by quantum chemical calculations, allowing us to unambiguously assign the absolute configurations of the first-eluted enantiomers as Rp in all of the cases examined. Interestingly, the CDs of 1 and 2, although largely comparable in shape, were still sensitive to the number and pattern of bromine substitution, showing closer resemblance between m-2 and o-2 and between p-2 and 1. The theoretical calculations also reproduced successfully these spectral resemblance between them. The anisotropy (g) factors for the 1Lb bands of these cyclophanes were considerably large (∼10–2), whereas those for the 1La band were conventional in the order of 10–3. In addition, a weak CE was observed in the low-energy region at around 320 nm, which turned out to originate from the interplanar interaction and is hence assigned to the “cyclophane band”. The experimental g factors of this band were fairly large in the order of 10–2, but the computation turned out to be quite challenging and were less well reproduced theoretically, ascribable to the forbidden nature of the transition.
@article{toda2018circular, title = {Circular Dichroisms of Mono-and Dibromo [2.2] paracyclophanes: A Combined Experimental and Theoretical Study}, author = {Toda, Mitsunobu and Inoue, Yoshihisa and Mori, Tadashi}, journal = {ACS Omega}, volume = {3}, issue = {1}, pages = {22--29}, year = {2018}, publisher = {ACS Publications}, doi = {10.1021/acsomega.7b01642}, url = {https://doi.org/10.1021/acsomega.7b01642}, dimensions = {true}, tab = {paper}, } -
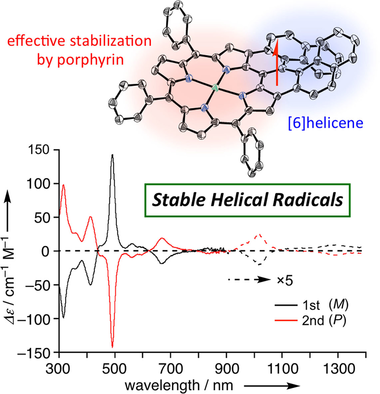 Porphyrin-Based Air-Stable Helical RadicalsKenichi Kato, Ko Furukawa*, Tadashi Mori, and Atsuhiro Osuka*Chem. Eur. J., 2018, 24, 572–575.
Porphyrin-Based Air-Stable Helical RadicalsKenichi Kato, Ko Furukawa*, Tadashi Mori, and Atsuhiro Osuka*Chem. Eur. J., 2018, 24, 572–575.Stable helical radicals are promising multi-functional molecules in light of intriguing magnetic and chiroptical properties. Attempts were made to extend diphenylmethyl-fused NiII porphyrin radical to helical system as the first air-stable organic neutral helical radicals. Intramolecular Pd-catalyzed twofold C−H arylation of methyl- or methoxy-introduced meso-diphenylmethyl NiII porphyrins gave a mixture of the target and rearranged radicals. Oxidative fusion reaction of meso-(bis(1-naphthyl)methyl) NiII porphyrins provided doubly fused NiII porphyrin radicals. One of the helical radicals was separated into enantiomers that showed mirror-image circular dichroism (CD) spectra up to 1300 nm. The helical dinaphthylmethyl-fused NiII porphyrin radical displayed solid-state magnetic property mostly arising from monomeric radicals, different from the parent diphenylmethyl-fused NiII porphyrin radical that showed antiferromagnetic coupling due to π-stacked pairing.
@article{kato2018porphyrin, title = {Porphyrin-Based Air-Stable Helical Radicals}, author = {Kato, Kenichi and Furukawa, Ko and Mori, Tadashi and Osuka, Atsuhiro}, journal = {Chem. Eur. J.}, volume = {24}, number = {3}, pages = {572--575}, year = {2018}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201705291}, url = {https://doi.org/10.1002/chem.201705291}, dimensions = {true}, tab = {paper}, } -
 Preface for special issue on photosynergeticsVasudevanpillai Biju*, Tadashi Mori*, Tsuyoshi Asahi*, and Hiroshi Miyasaka*J. Photochem. Photobiol. C: Photochem. Rev., 2018, 34, 1.
Preface for special issue on photosynergeticsVasudevanpillai Biju*, Tadashi Mori*, Tsuyoshi Asahi*, and Hiroshi Miyasaka*J. Photochem. Photobiol. C: Photochem. Rev., 2018, 34, 1.Molecules in the electronic excited state photo-energy conversion natural artificial photosynthesis so forth. For molecules in condensed phase however three general restrictions limit the efficient utilization of light energy. First molecules in higher electronically-excited states rapidly relax to the lowest excited electronic state (Kasha’s rule) extinguishing some portion of the absorbed photon energy. Second a large number of molecules excited in assemblies undergo fast annihilation leaving only a small fraction in the excited state leading to the loss of a major fraction of the number of photons absorbed by the assemblies. Third the electronic state accessible through one-photon absorption is limited by the optical selection rule denying access to various dark electronic excited states of molecules.
@article{biju2018preface, title = {Preface for special issue on photosynergetics}, author = {Biju, Vasudevanpillai and Mori, Tadashi and Asahi, Tsuyoshi and Miyasaka, Hiroshi}, journal = {J. Photochem. Photobiol. C: Photochem. Rev.}, volume = {34}, pages = {1}, year = {2018}, publisher = {Elsevier BV}, doi = {10.1016/j.jphotochemrev.2018.02.003.}, url = {https://doi.org/10.1016/j.jphotochemrev.2018.02.003.}, dimensions = {true}, tab = {paper}, } - Propeller ChiralityTadashi Mori*Kokagaku, 2018, 49, 163–166.
@article{mori2018propeller, title = {Propeller Chirality}, author = {Mori, Tadashi}, journal = {Kokagaku}, volume = {49}, issue = {3}, pages = {163--166}, year = {2018} }
2017
-
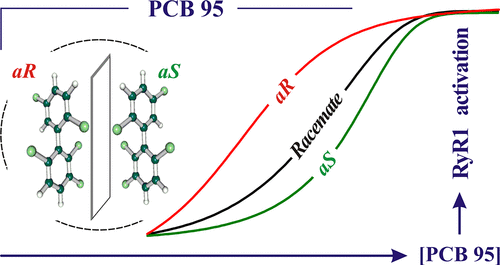 Enantioselectivity of 2, 2′, 3, 5′, 6-pentachlorobiphenyl (PCB 95) atropisomers toward ryanodine receptors (RyRs) and their influences on hippocampal neuronal networksWei Feng, Jing Zheng, Gaëlle Robin, Yao Dong, Makoto Ichikawa, Yoshihisa Inoue, Tadashi Mori, Takeshi Nakano, and Isaac N Pessah*Environ. Sci. Technol., 2017, 51, 14406–14416.
Enantioselectivity of 2, 2′, 3, 5′, 6-pentachlorobiphenyl (PCB 95) atropisomers toward ryanodine receptors (RyRs) and their influences on hippocampal neuronal networksWei Feng, Jing Zheng, Gaëlle Robin, Yao Dong, Makoto Ichikawa, Yoshihisa Inoue, Tadashi Mori, Takeshi Nakano, and Isaac N Pessah*Environ. Sci. Technol., 2017, 51, 14406–14416.Nineteen ortho-substituted PCBs are chiral and found enantioselectively enriched in ecosystems. Their differential actions on biological targets are not understood. PCB 95 (2,2′,3,5′,6-pentachlorobiphenyl), a chiral PCB of current environmental relevance, is among the most potent toward modifying ryanodine receptors (RyR) function and Ca2+ signaling. PCB 95 enantiomers are separated and assigned aR- and aS-PCB 95 using three chiral-column HPLC and circular dichroism spectroscopy. Studies of RyR1-enriched microsomes show aR-PCB 95 with >4× greater potency (EC50 = 0.20 ± 0.05 μM), ∼ 1.3× higher efficacy (Bmax = 3.74 ± 0.07 μM) in [3H]Ryanodine-binding and >3× greater rates (R = 7.72 ± 0.31 nmol/sec/mg) of Ca2+ efflux compared with aS-PCB 95, whereas racemate has intermediate activity. aR-PCB 95 has modest selectivity for RyR2, and lower potency than racemate toward the RyR isoform mixture in brain membranes. Chronic exposure of hippocampal neuronal networks to nanomolar PCB 95 during a critical developmental period shows divergent influences on synchronous Ca2+ oscillation (SCO): rac-PCB 95 increasing and aR-PCB 95 decreasing SCO frequency at 50 nM, although the latter’s effects are nonmonotonic at higher concentration. aS-PCB95 shows the greatest influence on inhibiting responses to 20 Hz electrical pulse trains. Considering persistence of PCB 95 in the environment, stereoselectivity toward RyRs and developing neuronal networks may clarify health risks associated with enantioisomeric enrichment of PCBs.
@article{feng2017enantioselectivity, title = {Enantioselectivity of 2, 2′, 3, 5′, 6-pentachlorobiphenyl (PCB 95) atropisomers toward ryanodine receptors (RyRs) and their influences on hippocampal neuronal networks}, author = {Feng, Wei and Zheng, Jing and Robin, Gaëlle and Dong, Yao and Ichikawa, Makoto and Inoue, Yoshihisa and Mori, Tadashi and Nakano, Takeshi and Pessah, Isaac N}, journal = {Environ. Sci. Technol.}, volume = {51}, issue = {24}, pages = {14406--14416}, year = {2017}, publisher = {ACS Publications}, doi = {10.1021/acs.est.7b04446}, url = {https://doi.org/10.1021/acs.est.7b04446}, dimensions = {true}, tab = {paper}, } -
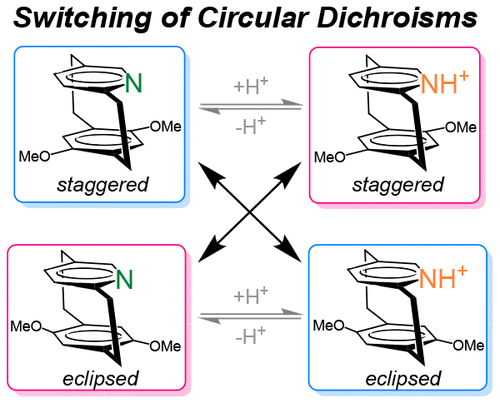 A combined experimental and theoretical study on the circular dichroism of staggered and eclipsed forms of dimethoxy [2.2]-,[3.2]-, and [3.3] pyridinophanes and their protonated formsAkinori Shimizu, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2017, 121, 8389–8398.
A combined experimental and theoretical study on the circular dichroism of staggered and eclipsed forms of dimethoxy [2.2]-,[3.2]-, and [3.3] pyridinophanes and their protonated formsAkinori Shimizu, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2017, 121, 8389–8398.The circular dichroisms (CDs) of dimethoxy[2.2]-, [3.2]-, and [3.3]pyridinophanes and their protonated forms were investigated experimentally and theoretically. Characteristic multisignate Cotton effects (CEs), typical for planar chiral cyclophane derivatives, were observed. The CD spectral pattern was quite comparable for the staggered forms of [2.2]-, [3.2]-, and [3.3]cyclophanes, but significantly differed for the eclipsed forms. More interestingly, the patterns resembled, but the CE signs were practically opposite between staggered and eclipsed [2.2]pyridinophanes. Upon protonation, the signs of most CEs were inverted in both forms of cyclophanes, due to the reversal of dipole moment in the pyridine against the pyridinium moiety. Such a change in CD spectrum upon protonation was not apparent in [3.2]pyridinophane, and the CD spectral behavior was more complex in [3.3]pyridinophanes. The variation of CD caused by the protonation/deprotonation process was temperature-dependent and hence utilized as a thermal sensor. The protonated forms of the homologous pyridinophanes with different tether lengths in staggered and eclipsed forms served as a model system for systematically studying the cation−π interaction and its effects on chiroptical properties. A steady increase of electronic interaction became apparent for the smaller-sized cyclophanes from the increased excitation energy and electronic coupling element of the charge-transfer (CT) band, while the observed CE at the CT band was a more complex function of the original transition dipole of donor/acceptor pair and linker atoms, as well as the strength of the electronic interaction.
@article{shimizu2017combined, title = {A combined experimental and theoretical study on the circular dichroism of staggered and eclipsed forms of dimethoxy [2.2]-,[3.2]-, and [3.3] pyridinophanes and their protonated forms}, author = {Shimizu, Akinori and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Phys. Chem. A}, volume = {121}, issue = {44}, pages = {8389--8398}, year = {2017}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.7b08623}, url = {https://doi.org/10.1021/acs.jpca.7b08623}, dimensions = {true}, tab = {paper}, } -
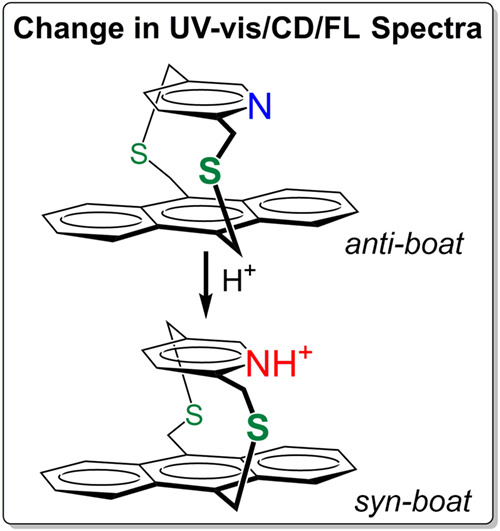 Chiroptical properties of dithia [3.3] cyclophanes composed of anthracene and pyridine/pyridinium moieties: A combined experimental and theoretical studyAkinori Shimizu, Keisuke Nagasaki, Yoshihisa Inoue, and Tadashi Mori*Chirality, 2017, 121, 8389–8398.
Chiroptical properties of dithia [3.3] cyclophanes composed of anthracene and pyridine/pyridinium moieties: A combined experimental and theoretical studyAkinori Shimizu, Keisuke Nagasaki, Yoshihisa Inoue, and Tadashi Mori*Chirality, 2017, 121, 8389–8398.Circular dichroisms (CDs) of neutral and protonated [3.3]anthracenopyridinophane (1 and 1-H+) were investigated experimentally and theoretically. Introducing an anthracene moiety with extended conjugation affected the cyclophane structure with the bent angles being appreciably reduced from those of parent [3.3]pyridinophane. The Cotton effects (CEs) observed at the 1Bb band for both 1 and 1-H+ were fairly strong and apparently bisignate, which, however, turned out not to be a simple exciton couplet but to be composed of multiple transitions. In contrast, the CEs were much weaker in the 1La band region. The spectral changes upon protonation were less significant compared with the parent pyridinophane, being dominated by the local transitions of anthracene. Nevertheless, the CD spectra of 1 and 1-H+ were well reproduced by theoretical calculations to allow us an unambiguous absolute configuration determination of the first high-performance liquid chromatography (HPLC) elute (from Chiralcel IB column) as Sp. The transannular interactions between the anthracene and pyridine/pyridinium units were examined by UV-vis and fluorescence spectroscopy to reveal a charge-transfer (CT) band in the low-energy region, particularly for 1-H+. Despite the comparable CT interactions, the CE at the CT band was much stronger for the anthracenopyridinophane than for the parent pyridinophane, affording an anisotropy (g) factor as large as 4 × 10-3.
@article{shimizu2017chiroptical, title = {Chiroptical properties of dithia [3.3] cyclophanes composed of anthracene and pyridine/pyridinium moieties: A combined experimental and theoretical study}, author = {Shimizu, Akinori and Nagasaki, Keisuke and Inoue, Yoshihisa and Mori, Tadashi}, journal = {Chirality}, volume = {121}, issue = {44}, pages = {8389--8398}, year = {2017}, publisher = {ACS Publications}, doi = {10.1002/chir.22740}, url = {https://doi.org/10.1002/chir.22740}, dimensions = {true}, tab = {paper}, } -
 Closed Pentaaza [9] helicene and Hexathia [9]/[5] helicene: Oxidative Fusion Reactions of ortho-Phenylene-Bridged Cyclic Hexapyrroles and HexathiophenesFengkun Chen, Takayuki Tanaka*, Yong Seok Hong, Tadashi Mori, Dongho Kim*, and Atsuhiro Osuka*Angew. Chem. Int. Ed., 2017, 129, 14880–14885.
Closed Pentaaza [9] helicene and Hexathia [9]/[5] helicene: Oxidative Fusion Reactions of ortho-Phenylene-Bridged Cyclic Hexapyrroles and HexathiophenesFengkun Chen, Takayuki Tanaka*, Yong Seok Hong, Tadashi Mori, Dongho Kim*, and Atsuhiro Osuka*Angew. Chem. Int. Ed., 2017, 129, 14880–14885.Oxidative fusion reactions of ortho-phenylene-bridged cyclic hexapyrroles and hexathiophenes furnished novel closed helicenes in a selective manner. X-Ray diffraction analysis unambiguously revealed the structures to be a closed pentaaza[9]helicene, the longest azahelicene reported so far, and an unexpected double-helical structure of hexathia[9]/[5]helicene, whose formation was assumed to result from multiple oxidative fusion along with a 1,2-aryl shift. The pentaaza[9]helicene exhibited well-defined emission with high fluorescence quantum yield (ΦF=0.31) among the known [9]helicenes. Chiral resolution of the racemic pentaaza[9]helicene and hexathia[9]/[5]helicene were achieved by chiral-phase HPLC and the enantiomers were characterized by circular dichroism spectra and DFT calculations.
@article{chen2017closed, title = {Closed Pentaaza [9] helicene and Hexathia [9]/[5] helicene: Oxidative Fusion Reactions of ortho-Phenylene-Bridged Cyclic Hexapyrroles and Hexathiophenes}, author = {Chen, Fengkun and Tanaka, Takayuki and Hong, Yong Seok and Mori, Tadashi and Kim, Dongho and Osuka, Atsuhiro}, journal = {Angew. Chem. Int. Ed.}, volume = {129}, issue = {46}, pages = {14880--14885}, year = {2017}, publisher = {Wiley Online Library}, doi = {10.1002/anie.201708429}, url = {https://doi.org/10.1002/anie.201708429}, dimensions = {true}, tab = {paper}, } -
 Intense redox-driven chiroptical switching with a 580 mV hysteresis actuated through reversible dimerization of an azoniaheliceneJochen R Brandt, Lubomír Pospíšil, Lucie Bednárová, Rosenildo Correa Costa, Andrew JP White, Tadashi Mori, Filip Teplý, and Matthew J Fuchter*Chem. Commun., 2017, 53, 9059–9062.
Intense redox-driven chiroptical switching with a 580 mV hysteresis actuated through reversible dimerization of an azoniaheliceneJochen R Brandt, Lubomír Pospíšil, Lucie Bednárová, Rosenildo Correa Costa, Andrew JP White, Tadashi Mori, Filip Teplý, and Matthew J Fuchter*Chem. Commun., 2017, 53, 9059–9062.Electrochemical reduction of an azoniahelicene affords a dimer, accompanied by a strong change in the electronic circular dichroism. The fast dimerisation event leads to a >500 mV shift of the oxidation potential, affording a large area of bistability, where the chiroptical signal only depends on the redox history.
@article{brandt2017intense, title = {Intense redox-driven chiroptical switching with a 580 mV hysteresis actuated through reversible dimerization of an azoniahelicene}, author = {Brandt, Jochen R and Pospíšil, Lubomír and Bednárová, Lucie and da Costa, Rosenildo Correa and White, Andrew JP and Mori, Tadashi and Teplý, Filip and Fuchter, Matthew J}, journal = {Chem. Commun.}, volume = {53}, issue = {65}, pages = {9059--9062}, year = {2017}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c7cc04903j}, url = {https://doi.org/10.1039/c7cc04903j}, dimensions = {true}, tab = {paper}, } -
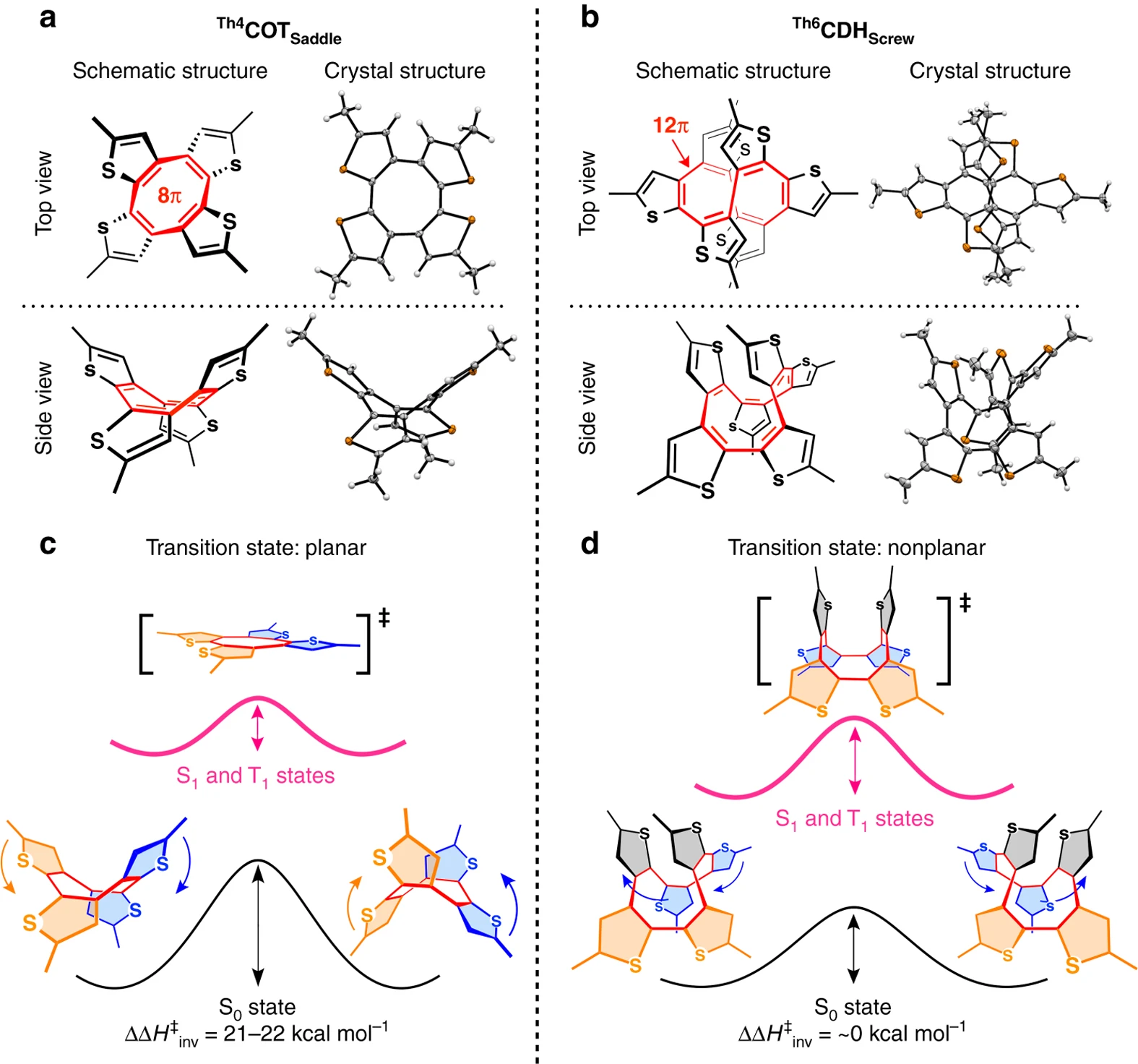 Energetics of Baird aromaticity supported by inversion of photoexcited chiral [4n] annulene derivativesMichihisa Ueda, Kjell Jorner, Young Mo Sung, Tadashi Mori, Qi Xiao, Dongho Kim, Henrik Ottosson, Takuzo Aida, and Yoshimitsu Itoh*Nat. Commun., 2017, 8, 346.
Energetics of Baird aromaticity supported by inversion of photoexcited chiral [4n] annulene derivativesMichihisa Ueda, Kjell Jorner, Young Mo Sung, Tadashi Mori, Qi Xiao, Dongho Kim, Henrik Ottosson, Takuzo Aida, and Yoshimitsu Itoh*Nat. Commun., 2017, 8, 346.For the concept of aromaticity, energetic quantification is crucial. However, this has been elusive for excited-state (Baird) aromaticity. Here we report our serendipitous discovery of two nonplanar thiophene-fused chiral [4n]annulenes Th4 COT Saddle and Th6 CDH Screw , which by computational analysis turned out to be a pair of molecules suitable for energetic quantification of Baird aromaticity. Their enantiomers were separable chromatographically but racemized thermally, enabling investigation of the ring inversion kinetics. In contrast to Th6 CDH Screw , which inverts through a nonplanar transition state, the inversion of Th4 COT Saddle , progressing through a planar transition state, was remarkably accelerated upon photoexcitation. As predicted by Baird’s theory, the planar conformation of Th4 COT Saddle is stabilized in the photoexcited state, thereby enabling lower activation enthalpy than that in the ground state. The lowering of the activation enthalpy, i.e., the energetic impact of excited-state aromaticity, was quantified experimentally to be as high as 21–22 kcal mol–1.
@article{ueda2017energetics, title = {Energetics of Baird aromaticity supported by inversion of photoexcited chiral [4n] annulene derivatives}, author = {Ueda, Michihisa and Jorner, Kjell and Sung, Young Mo and Mori, Tadashi and Xiao, Qi and Kim, Dongho and Ottosson, Henrik and Aida, Takuzo and Itoh, Yoshimitsu}, journal = {Nat. Commun.}, volume = {8}, issue = {1}, pages = {346}, year = {2017}, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s41467-017-00382-1}, url = {https://doi.org/10.1038/s41467-017-00382-1}, dimensions = {true}, tab = {paper}, } -
 Oligosaccharide Sensing in Aqueous Media by Porphyrin–Curdlan Conjugates: A Prêt-á-Porter Rather Than Haute-Couture ApproachGaku Fukuhara*, Mayuko Sasaki, Munenori Numata, Tadashi Mori, and Yoshihisa Inoue*Chem. Eur. J., 2017, 23, 11272–11278.
Oligosaccharide Sensing in Aqueous Media by Porphyrin–Curdlan Conjugates: A Prêt-á-Porter Rather Than Haute-Couture ApproachGaku Fukuhara*, Mayuko Sasaki, Munenori Numata, Tadashi Mori, and Yoshihisa Inoue*Chem. Eur. J., 2017, 23, 11272–11278.Saccharide sensing in aqueous media is an intriguing but challenging goal in current chemistry. Herein we report the oligosaccharide-sensing behavior of newly synthesized porphyrin–curdlan conjugates, which are random coils in DMSO but become globules in aqueous solutions to induce circular dichroism (ICD) in the biologically accessible spectral region due to the conformational fixation of porphyrin reporters. The magnitude of ICD was significantly varied specifically in the presence of acarbose, a drug for type-2 diabetes, enabling us to detect the aminosaccharide at concentrations down to 200 μm. This result demonstrates that the prêt-á-porter approach, using less-defined reporter-curdlan conjugates, is more advantageous than the traditional haute-couture approach with highly sophisticated hosts in particular in oligosaccharide sensing.
@article{fukuhara2017oligosaccharide, title = {Oligosaccharide Sensing in Aqueous Media by Porphyrin--Curdlan Conjugates: A Prêt-á-Porter Rather Than Haute-Couture Approach}, author = {Fukuhara, Gaku and Sasaki, Mayuko and Numata, Munenori and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chem. Eur. J.}, volume = {23}, issue = {47}, pages = {11272--11278}, year = {2017}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201701360}, url = {https://doi.org/10.1002/chem.201701360}, dimensions = {true}, tab = {paper}, } -
 Temperature-driven planar chirality switching of a pillar [5] arene-based molecular universal jointJiabin Yao, Wanhua Wu, Wenting Liang, Yujun Feng, Dayang Zhou, Jason J Chruma, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue, and Cheng Yang*Angew. Chem. Int. Ed., 2017, 129, 6973–6977.
Temperature-driven planar chirality switching of a pillar [5] arene-based molecular universal jointJiabin Yao, Wanhua Wu, Wenting Liang, Yujun Feng, Dayang Zhou, Jason J Chruma, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue, and Cheng Yang*Angew. Chem. Int. Ed., 2017, 129, 6973–6977.The study of an enantiopure bicyclic pillar[5]arene-based molecular universal joint (MUJ) by single-crystal X-ray diffraction allowed for the first time the unequivocal assignment of the absolute configuration of a planar chiral pillar[5]arene by circular dichroism spectroscopy. Crucially, the absolute configuration of the MUJ was switched reversibly by temperature, with an accompanying sign inversion of the anisotropy factor that varied by as much as 0.03, which is the largest value ever reported. Mechanistically, the reversible chirality switching of the MUJ is driven by the threading/dethreading motion of the fused ring and hence is dependent on both the size and nature of the ring and the solvent employed, reflecting the critical balance between the self-complexation of the ring by pillar[5]arene, the solvation to the excluded ring, and the inclusion of solvent molecules in the cavity.
@article{yao2017temperature, title = {Temperature-driven planar chirality switching of a pillar [5] arene-based molecular universal joint}, author = {Yao, Jiabin and Wu, Wanhua and Liang, Wenting and Feng, Yujun and Zhou, Dayang and Chruma, Jason J and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa and Yang, Cheng}, journal = {Angew. Chem. Int. Ed.}, volume = {129}, issue = {24}, pages = {6973--6977}, year = {2017}, publisher = {Wiley Online Library}, doi = {10.1002/anie.201702542}, url = {https://doi.org/10.1002/anie.201702542}, dimensions = {true}, tab = {paper}, } -
 Absolute configuration determination through the unique intramolecular excitonic coupling in the circular dichroisms of o, p′-DDT and o, p′-DDD. A combined experimental and theoretical studyHiroki Tanaka, Yoshihisa Inoue, Takeshi Nakano, and Tadashi Mori*Photochem. Photobiol. Sci., 2017, 16, 606–610.
Absolute configuration determination through the unique intramolecular excitonic coupling in the circular dichroisms of o, p′-DDT and o, p′-DDD. A combined experimental and theoretical studyHiroki Tanaka, Yoshihisa Inoue, Takeshi Nakano, and Tadashi Mori*Photochem. Photobiol. Sci., 2017, 16, 606–610.Circular dichroisms (CDs) of the o,p′-isomers of 1,1,1-trichloro- and 1,1-dichloro-2,2-bis(chlorophenyl)ethanes (DDT and DDD) were investigated experimentally and theoretically. A series of strong Cotton effect peaks in a characteristic negative–negative–positive–negative, or its mirror-imaged, pattern were observed in the CD spectra of these persistent organic pollutants. The theoretical CD spectra at the SAC-CI/B95(d) and RI-CC2/def2-TZVPP levels well reproduced the experimental ones, enabling us to unambiguously assign the absolute configuration of (+)-DDT and (−)-DDD as S.
@article{tanaka2017absolute, title = {Absolute configuration determination through the unique intramolecular excitonic coupling in the circular dichroisms of o, p′-DDT and o, p′-DDD. A combined experimental and theoretical study}, author = {Tanaka, Hiroki and Inoue, Yoshihisa and Nakano, Takeshi and Mori, Tadashi}, journal = {Photochem. Photobiol. Sci.}, volume = {16}, issue = {4}, pages = {606--610}, year = {2017}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c6pp00438e}, url = {https://doi.org/10.1039/c6pp00438e}, dimensions = {true}, tab = {paper}, } -
 Möbius aromatic [28] hexaphyrin germanium (IV) and tin (IV) complexes: Efficient formation of triplet excited statesMondo Izawa, Taeyeon Kim, Shin-ichiro Ishida, Takayuki Tanaka, Tadashi Mori, Dongho Kim*, and Atsuhiro Osuka*Angew. Chem. Int. Ed., 2017, 129, 4040–4044.
Möbius aromatic [28] hexaphyrin germanium (IV) and tin (IV) complexes: Efficient formation of triplet excited statesMondo Izawa, Taeyeon Kim, Shin-ichiro Ishida, Takayuki Tanaka, Tadashi Mori, Dongho Kim*, and Atsuhiro Osuka*Angew. Chem. Int. Ed., 2017, 129, 4040–4044.[28]Hexaphyrin GeIV and SnIV complexes were synthesized in high yields by reactions of [28]hexaphyrin with GeCl4 or SnCl4 in the presence of triethylamine. Both complexes display distinct 28π Möbius aromatic character and possess a trigonal bipyramidal geometry at the central GeIV or SnIV atom. The equatorial hydroxy group of the GeIV complex was smoothly exchanged with neutral nucleophiles, such as phenol derivatives and thiophenol, with retention of configuration. In the SnIV complex, intersystem crossing to the T1 state is remarkably enhanced owing to the effective heavy-atom effect, thus allowing the formation of the T1 tate in high yield. The T1 states of the GeIV and SnVI complexes were found to be antiaromatic on the basis of the transient absorption features in line with the Baird rule.
@article{izawa2017mobius, title = {Möbius aromatic [28] hexaphyrin germanium (IV) and tin (IV) complexes: Efficient formation of triplet excited states}, author = {Izawa, Mondo and Kim, Taeyeon and Ishida, Shin-ichiro and Tanaka, Takayuki and Mori, Tadashi and Kim, Dongho and Osuka, Atsuhiro}, journal = {Angew. Chem. Int. Ed.}, volume = {129}, issue = {14}, pages = {4040--4044}, year = {2017}, publisher = {Wiley Online Library}, doi = {10.1002/anie.201700063}, url = {https://doi.org/10.1002/anie.201700063}, dimensions = {true}, tab = {paper}, } -
 Sui generis helicene-based supramolecular chirogenic system: Enantioselective sensing, solvent control, and application in chiral group transfer reactionMohammed Hasan, Vaibhav N Khose, Tadashi Mori, Victor Borovkov*, and Anil V Karnik*ACS Omega, 2017, 2, 592–598.
Sui generis helicene-based supramolecular chirogenic system: Enantioselective sensing, solvent control, and application in chiral group transfer reactionMohammed Hasan, Vaibhav N Khose, Tadashi Mori, Victor Borovkov*, and Anil V Karnik*ACS Omega, 2017, 2, 592–598.A novel dioxa[6]helicene-based supramolecular chirogenic system (1) as a specific chiral recognition host for enantiopure trans-1,2-cyclohexanediamine (2) is reported. Host 1 with an inherent free phenolic group and a (1S)-camphanate chiral handle on the opposite terminal rings of the helicene chromophore acted as an efficient turn on fluorescent sensor for S,S-2 with an excellent enantioselective factor, α = KSS/KRR = 6.3 in benzene. This specific host–guest interaction phenomenon is found to be solvent-dependent, which leads to an enantioselective chiral (camphanate) group transfer to the diamine guest molecule. In the case of R,R-2, the de value is up to 68% even at room temperature. Intriguingly, the induced helicity in dioxa[6]helicene diol 6, upon supramolecular hydrogen-bonding interactions, is of opposite sense with positive helicity for S,S-2 and negative helicity for R,R-2, as shown by circular dichroism spectroscopy and in combination with theoretical calculations. This chiral supramolecular system is found to be an excellent host–guest pair for enantiomeric recognition of 2, based on their electronic and steric factors.
@article{hasan2017sui, title = {Sui generis helicene-based supramolecular chirogenic system: Enantioselective sensing, solvent control, and application in chiral group transfer reaction}, author = {Hasan, Mohammed and Khose, Vaibhav N and Mori, Tadashi and Borovkov, Victor and Karnik, Anil V}, journal = {ACS Omega}, volume = {2}, issue = {2}, pages = {592--598}, year = {2017}, publisher = {ACS Publications}, doi = {10.1021/acsomega.6b00522}, url = {https://doi.org/10.1021/acsomega.6b00522}, dimensions = {true}, tab = {paper}, } -
 Temperature Dynamics of Single Molecular Antifreeze ProteinRio Okada, Tatsuya Arai, Daichi Fukami, Yuhuku Matsushita, Jae-won Chang, Hiroshi Sekiguchi, Noboru Ohta, Tadashi Mori, Masaki Nishijima, Keisuke Miyazawa, Takeshi Fukuma, Keigo Ikezaki, Sakae Tsuda, and Yuji C SasakiBiophysical Journal, 2017, 112, 323a.
Temperature Dynamics of Single Molecular Antifreeze ProteinRio Okada, Tatsuya Arai, Daichi Fukami, Yuhuku Matsushita, Jae-won Chang, Hiroshi Sekiguchi, Noboru Ohta, Tadashi Mori, Masaki Nishijima, Keisuke Miyazawa, Takeshi Fukuma, Keigo Ikezaki, Sakae Tsuda, and Yuji C SasakiBiophysical Journal, 2017, 112, 323a.AntiFreeze Proteins (=AFPs) bind to a surface of ice crystals and inhibit their growth. Some living organisms; fishes, insects and fungus, at low-temperature environment have several different types of AFPs. AFPs protect their body from freezing damages. To clarify the antifreeze effect of lpAFP (isolated from longsnout poacher, a fish living in the sea of Okhotsk) at single molecular level, we performed the single molecular observation using diffracted X-ray tracking (=DXT). DXT is the method to observe single molecular motions using X-rays and gold nanocrystals as probe. Gold nanocrystals are labeled on AFPs, and when irradiated with X-rays, diffracted spots from gold nanocrystals can be observed. We can observe the motion of the AFPs by tracking these spots. We performed the DXT experiments at SPring-8 BL40XU, and the spatial and time resolutions are the order of milli-radian and micro-second. In this study, we used lpAFP isolated from longsnout poacher, and observed the temperature dependency of the lpAFP’s single molecular brownian motion. As a result, lpAFP’s motion increased at 5 °C. Next, to ensure the relationship between the adsorption property and the maximum brownian motion, we observed the temperature dependency of the adsorption affinity to AgI thin film as ice surface. As a result, the adsorption amount to AgI of lpAFP was the largest at 2°C. These two experiments suggest that there is a strong correlation between lpAFP’s molecular motion and adsorption property. We acquired the temperature dependency of lpAFP using other experiments; circular dichroism (CD), atomic force microscopy (AFM), dynamic light scattering (DLS), and small angle X-ray scattering (SAXS, performed at SPring-8 BL40B2). In poster session, we discuss the detail experimental procedure and results.
@article{okada2017temperature, title = {Temperature Dynamics of Single Molecular Antifreeze Protein}, author = {Okada, Rio and Arai, Tatsuya and Fukami, Daichi and Matsushita, Yuhuku and Chang, Jae-won and Sekiguchi, Hiroshi and Ohta, Noboru and Mori, Tadashi and Nishijima, Masaki and Miyazawa, Keisuke and Fukuma, Takeshi and Ikezaki, Keigo and Tsuda, Sakae and Sasaki, Yuji C}, journal = {Biophysical Journal}, volume = {112}, issue = {3}, pages = {323a}, year = {2017}, publisher = {Elsevier}, doi = {10.1016/j.bpj.2016.11.1751}, url = {https://doi.org/10.1016/j.bpj.2016.11.1751}, dimensions = {true}, tab = {paper}, } -
 Protonation-induced sign inversion of the cotton effects of pyridinophanes. A combined experimental and theoretical studyAkinori Shimizu, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2017, 121, 977–985.
Protonation-induced sign inversion of the cotton effects of pyridinophanes. A combined experimental and theoretical studyAkinori Shimizu, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2017, 121, 977–985.The circular dichroisms (CDs) of planar chiral [2.2]- and [3.3]-pyridinophanes were investigated experimentally and theoretically. Strong multisignate Cotton effects, typical for cyclophane derivatives, were observed. The CD spectra of [2.2]- and [3.3]-paracyclophanes closely resembled in pattern each other, despite the much greater conformational variations in the latter. Upon protonation, both of the cyclophanes suffered dramatic CD spectral changes with accompanying complete sign inversion, which was attributed to the reversal of diploe moment of pyridinium versus pyridine moiety. This chiroptical property switching driven by protonation/deprotonation was temperature-dependent and hence applicable to thermal sensing. The protonated forms of pyridinophanes served as ideal model systems for studying the cation−π interactions and their effects on chiroptical properties. Thus, the molar CD (Δε) of the charge-transfer band of protonated [2.2]pyridinophane was 10-fold larger than that of protonated [3.3]pyridinophane, which exceeds the increased interplane electronic interactions assessed from the electronic coupling element values.
@article{shimizu2017protonation, title = {Protonation-induced sign inversion of the cotton effects of pyridinophanes. A combined experimental and theoretical study}, author = {Shimizu, Akinori and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Phys. Chem. A}, volume = {121}, issue = {5}, pages = {977--985}, year = {2017}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.6b12287}, url = {https://doi.org/10.1021/acs.jpca.6b12287}, dimensions = {true}, tab = {paper}, } -
 Propeller chirality of boron heptaaryldipyrromethene: unprecedented supramolecular dimerization and chiroptical propertiesMasataka Toyoda, Yoshitane Imai, and Tadashi Mori*J. Phys. Chem. Lett., 2017, 8, 42–48.
Propeller chirality of boron heptaaryldipyrromethene: unprecedented supramolecular dimerization and chiroptical propertiesMasataka Toyoda, Yoshitane Imai, and Tadashi Mori*J. Phys. Chem. Lett., 2017, 8, 42–48.Chiral boron dipyrromethenes (BPs) enjoy high fluorescence efficiency at visible to near-IR wavelength regions with a reasonable range of dissymmetry factors. Here, we demonstrate that the (quasi)propeller chirality, similarly to hexagonal propeller in hexaarylbenzene, can be effectively induced in heptaarylated BP. In addition, supramolecular dimer was formed at low temperatures in nonpolar solvent, which exhibits strong bisignate Cotton effects at BP transitions (the couplet amplitude A = 193 M–1 cm–1) in the circular dichroism (CD). Due to the bulky substituents on the propeller blades, but with void space around boron atoms, BP chromophores in the dimer are aligned in a head-to-tail manner with a small torsion (φ ≈ 15°), to avoid fluorescence quenching usually observed in H-type dimer of BPs, exhibiting strong circularly polarized luminescence (CPL) signals (glum = 2.0 × 10–3, Φlum = 0.45). Such supramolecular dimer formation would be viewed as an alternative approach for designing and developing novel chiroptical materials.
@article{toyoda2017propeller, title = {Propeller chirality of boron heptaaryldipyrromethene: unprecedented supramolecular dimerization and chiroptical properties}, author = {Toyoda, Masataka and Imai, Yoshitane and Mori, Tadashi}, journal = {J. Phys. Chem. Lett.}, volume = {8}, issue = {1}, pages = {42--48}, year = {2017}, publisher = {ACS Publications}, doi = {10.1021/acs.jpclett.6b02492}, url = {https://doi.org/10.1021/acs.jpclett.6b02492}, dimensions = {true}, tab = {paper}, } -
 Asymmetric Photochemical Synthesis Based on Selective Excitation of Charge-Transfer ComplexesTadashi Mori*J. Syn. Org. Chem., Jpn., 2017, 75, 144–152.
Asymmetric Photochemical Synthesis Based on Selective Excitation of Charge-Transfer ComplexesTadashi Mori*J. Syn. Org. Chem., Jpn., 2017, 75, 144–152.The use of photoreactions such as photoredox catalysts in asymmetric synthesis has been expansively recognized as expedient procedure over the past decade. In this paper, a potential application of photoreaction of Charge-Transfer (CT) complex for asymmetric synthesis is described. In chiral donor-acceptor systems, excitation wavelength, along with other environmental factors such as solvent polarity and temperature, plays more substantial role in determining the stereoselectivity of the photochemical outcomes. Accordingly, the excited CT complex produced upon the selective CT-band excitation behaves completely different from the conventional exciplex, which is formed by the direct excitation of donor or acceptor. Such unique wavelength effect, together with other entropic controls, may possibly come to be valuable means in asymmetric photochemical synthesis for the future.
@article{mori2017asymmetric, title = {Asymmetric Photochemical Synthesis Based on Selective Excitation of Charge-Transfer Complexes}, author = {Mori, Tadashi}, journal = {J. Syn. Org. Chem., Jpn.}, volume = {75}, number = {2}, pages = {144--152}, year = {2017}, doi = {10.5059/yukigoseikyokaishi.75.144}, url = {https://doi.org/10.5059/yukigoseikyokaishi.75.144}, dimensions = {true}, tab = {review}, } - Entropy Control of ReactionsTadashi MoriJ. Syn. Org. Chem., Jpn., 2017, 75, 160.
@article{mori2017entropy, title = {Entropy Control of Reactions}, author = {Mori, Tadashi}, journal = {J. Syn. Org. Chem., Jpn.}, volume = {75}, issue = {2}, pages = {160}, year = {2017}, doi = {10.5059/yukigoseikyokaishi.75.160}, url = {https://doi.org/10.5059/yukigoseikyokaishi.75.160}, dimensions = {true}, tab = {review} } - Chiral Photochemistry of Charge Transfer Complexes: Effect of Excitation WavelengthTadashi Mori*Symmetry, 2017, 28, 207–210.
@article{mori2017chiral, title = {Chiral Photochemistry of Charge Transfer Complexes: Effect of Excitation Wavelength}, author = {Mori, Tadashi}, journal = {Symmetry}, volume = {28}, issue = {2}, pages = {207--210}, year = {2017}, publisher = {Symmetry: Culture and Science} }
2016
-
 Highly enantiodifferentiating site of human serum albumin for mediating photocyclodimerization of 2-anthracenecarboxylate elucidated by site-specific inhibition/quenching with xenonMasaki Nishijima*, Tamara CS Pace, Cornelia Bohne, Tadashi Mori, Yoshihisa Inoue, and Takehiko WadaJ. Photochem. Photobiol. A: Chem., 2016, 331, 89–94.
Highly enantiodifferentiating site of human serum albumin for mediating photocyclodimerization of 2-anthracenecarboxylate elucidated by site-specific inhibition/quenching with xenonMasaki Nishijima*, Tamara CS Pace, Cornelia Bohne, Tadashi Mori, Yoshihisa Inoue, and Takehiko WadaJ. Photochem. Photobiol. A: Chem., 2016, 331, 89–94.Complementary to the previous assignment of the first, second, and fourth binding sites of human serum albumin (HSA) for 2-anthracenecarboxylate (AC) and the subsequent mediation of AC photocyclodimerization, the site-specific inhibition of the enantiodifferentiation by xenon allowed us to assign the remaining third and fifth AC-binding sites to subdomains IB and IIIB, respectively. This study reveals a clearer picture of the binding and photochirogenic behavior of HSA and further expands the scope of bio-supramolecular photochirogenesis.
@article{nishijima2016highly, title = {Highly enantiodifferentiating site of human serum albumin for mediating photocyclodimerization of 2-anthracenecarboxylate elucidated by site-specific inhibition/quenching with xenon}, author = {Nishijima, Masaki and Pace, Tamara CS and Bohne, Cornelia and Mori, Tadashi and Inoue, Yoshihisa and Wada, Takehiko}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {331}, pages = {89--94}, year = {2016}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2015.12.019}, url = {https://doi.org/10.1016/j.jphotochem.2015.12.019}, dimensions = {true}, tab = {paper}, } -
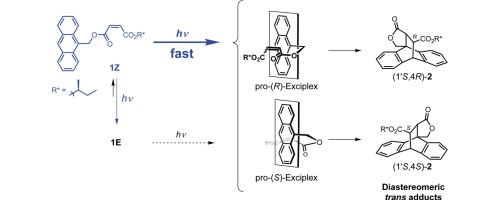 Enhanced asymmetric photocycloaddition of anthracene tethered to maleate versus fumarate through non-fluorescent exciplex intermediateMakoto Ichikawa, Yoshihisa Inoue, and Tadashi Mori*J. Photochem. Photobiol. A: Chem., 2016, 331, 102–109.
Enhanced asymmetric photocycloaddition of anthracene tethered to maleate versus fumarate through non-fluorescent exciplex intermediateMakoto Ichikawa, Yoshihisa Inoue, and Tadashi Mori*J. Photochem. Photobiol. A: Chem., 2016, 331, 102–109.The asymmetric photocycloaddition of anthracene-tethered maleate with a small (S)-1-methylpropyl auxiliary at the peripheral position (1Z) underwent much faster than that of the corresponding fumarate (1E). The absolute configuration of the newly created chiral center in the major product was ambiguously assigned as (4S) by X-ray crystallographic as well as CD spectral studies. The facile E–Z photoisomerization was simultaneously observed for both 1Z and 1E, again much faster for 1Z. The concurrent electron transfer works as a quenching process and the fumarate acted as a better quencher than maleate in both intramolecular and intermolecular systems. The fluorescence quenching and quantum yield investigations also supported such facile electron transfer from excited-state anthracene to acceptor in the singlet manifold. Observed diastereoselectivities in the photocycloaddition of 1Z and 1E were moderate but substantial (5–12% de), given the fact that the chiral auxiliary is introduced at the remote position from the reaction center (4-bond separation). The non-fluorescent exciplex, probably in a triplet manifold derived from the singlet exciplex, plays a crucial role in this efficient cyclization process, affording a biradical intermediate, that can either dissociate or undergo second bond formation to afford a diastereomeric trans adduct 2. The diastereoselectivity was higher in less polar solvents, suggesting the polar nature of the exciplex.
@article{ichikawa2016enhanced, title = {Enhanced asymmetric photocycloaddition of anthracene tethered to maleate versus fumarate through non-fluorescent exciplex intermediate}, author = {Ichikawa, Makoto and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {331}, pages = {102--109}, year = {2016}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2015.09.001}, url = {https://doi.org/10.1016/j.jphotochem.2015.09.001}, dimensions = {true}, tab = {paper}, } -
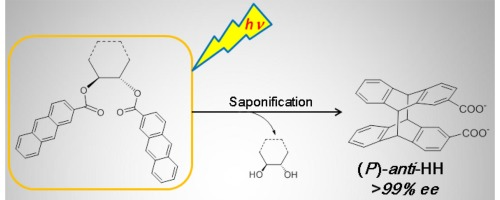 Critical control by scaffold flexibility achieved in diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylateGaku Fukuhara*, Kazuhiro Iida, Tadashi Mori, and Yoshihisa InoueJ. Photochem. Photobiol. A: Chem., 2016, 331, 76–83.
Critical control by scaffold flexibility achieved in diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylateGaku Fukuhara*, Kazuhiro Iida, Tadashi Mori, and Yoshihisa InoueJ. Photochem. Photobiol. A: Chem., 2016, 331, 76–83.By progressively increasing the flexibility of chiral vicinal diol scaffold (from rigid cyclic tetrasaccharide to flexible 2,3-butanediol via glucose and trans-1,2-cyclohexandiol) in the diastereodifferentiating photocyclodimerization to head-to-head (HH) dimers of 2-anthracenecarboxylate on the scaffold, the anti/syn preference was dramatically inverted from 42:1 to 1:12, while the enantiomeric excess of the chiral anti-HH dimer was consistently kept high at >99% due to the excited-state dynamics that strongly disfavors the si–si enantiotopic face attack against the antipodal re–re face attack, exclusively affording the (P)-enantiomer.
@article{fukuhara2016critical, title = {Critical control by scaffold flexibility achieved in diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate}, author = {Fukuhara, Gaku and Iida, Kazuhiro and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {331}, pages = {76--83}, year = {2016}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2016.01.016}, url = {https://doi.org/10.1016/j.jphotochem.2016.01.016}, dimensions = {true}, tab = {paper}, } -
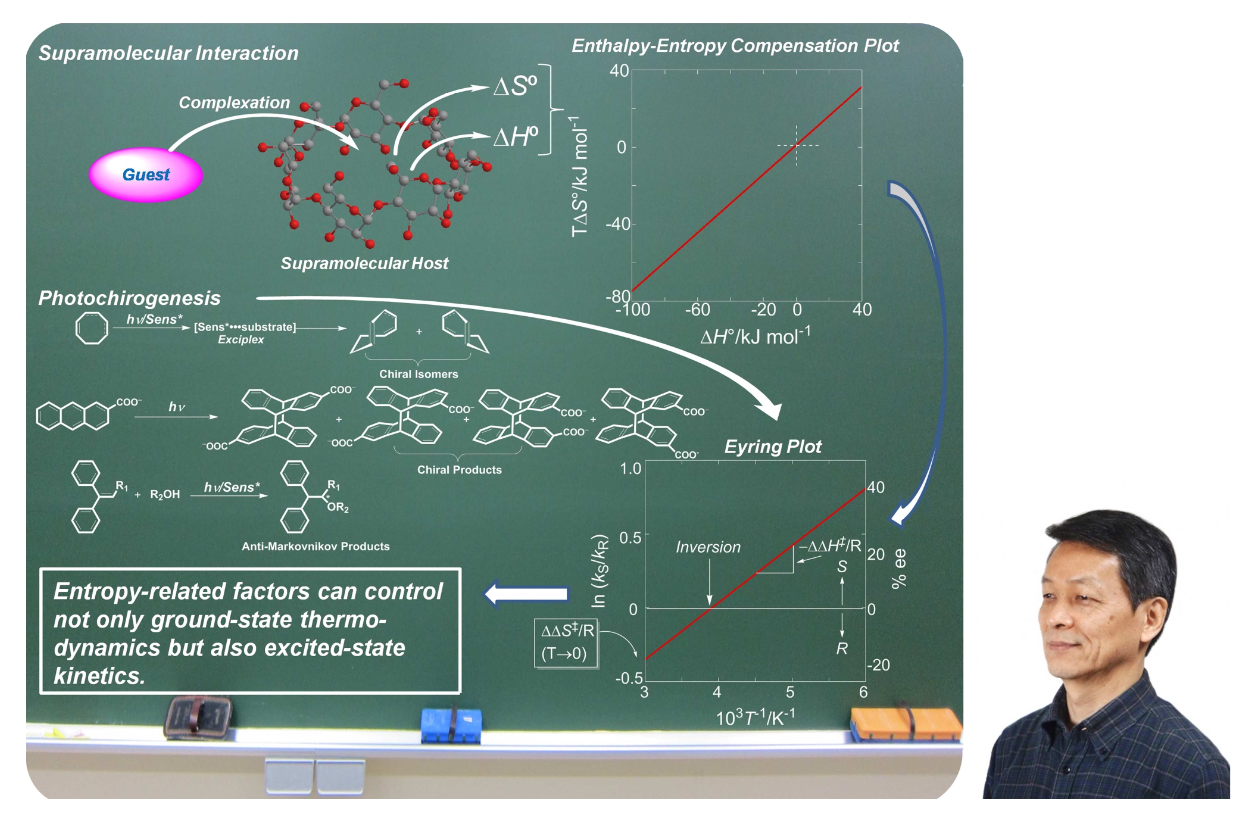 Yoshihisa Inoue—A researcher’s quest for photochirogenesisTadashi Mori*, Gaku Fukuhara, and Takehiko WadaJ. Photochem. Photobiol. A: Chem., 2016, 331, 2–7.
Yoshihisa Inoue—A researcher’s quest for photochirogenesisTadashi Mori*, Gaku Fukuhara, and Takehiko WadaJ. Photochem. Photobiol. A: Chem., 2016, 331, 2–7.This issue of Journal of Photochemistry and Photobiology A: Chemistry is dedicated to Prof. Yoshihisa Inoue, a man of “photochirogenesis”, and his enthusiastic attitude toward chemistry. We feel highly privileged and deeply honored that we have been serving as guest editors for this special issue, honoring his retirement from Osaka University in 2015 and continued impact on supramolecular chemistry and photochemistry.
@article{mori2016yoshihisa, title = {Yoshihisa Inoue—A researcher’s quest for photochirogenesis}, author = {Mori, Tadashi and Fukuhara, Gaku and Wada, Takehiko}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {331}, pages = {2--7}, year = {2016}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2016.08.006}, url = {https://doi.org/10.1016/j.jphotochem.2016.08.006}, dimensions = {true}, tab = {paper}, } -
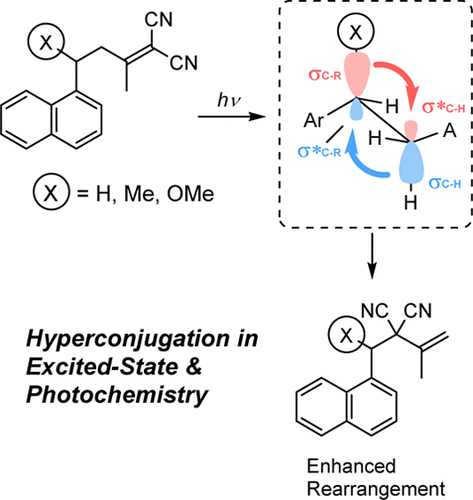 Orbital Control of Photochemical Rearrangement of 4-Aryl-1, 1-dicyano-1-butenes through the Hyperconjugative Substitution on the Linker ChainNobuo Matsuki, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. Lett., 2016, 7, 4957–4961.
Orbital Control of Photochemical Rearrangement of 4-Aryl-1, 1-dicyano-1-butenes through the Hyperconjugative Substitution on the Linker ChainNobuo Matsuki, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. Lett., 2016, 7, 4957–4961.Hyperconjugative interaction was demonstrated to play a vital role in the photochemistry of 4-aryl-1,1-dicyano-1-butenes. Thus a simple substituent on the benzylic position effectively induced a new photoreactivity to afford an allylic rearrangement product that is not obtained for the parent substrate. The natural bond orbital analysis was employed to reveal the enhanced relative contributions of hyperconjugation in the excited state, which dramatically alter the photochemical outcomes not only by reducing the strength of the allylic/benzylic bond but more crucially by affecting the conformer distribution.
@article{matsuki2016orbital, title = {Orbital Control of Photochemical Rearrangement of 4-Aryl-1, 1-dicyano-1-butenes through the Hyperconjugative Substitution on the Linker Chain}, author = {Matsuki, Nobuo and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Phys. Chem. Lett.}, volume = {7}, issue = {24}, pages = {4957--4961}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/acs.jpclett.6b02632}, url = {https://doi.org/10.1021/acs.jpclett.6b02632}, dimensions = {true}, tab = {paper}, } -
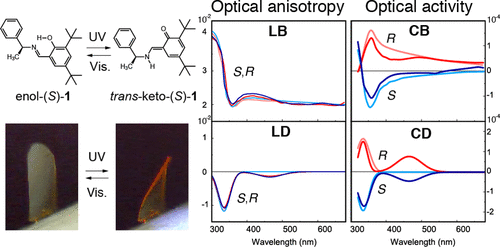 Optical activity and optical anisotropy in photomechanical crystals of chiral salicylidenephenylethylaminesAkifumi Takanabe, Masahito Tanaka, Kohei Johmoto, Hidehiro Uekusa, Tadashi Mori, Hideko Koshima*, and Toru Asahi*J. Am. Chem. Soc., 2016, 138, 15066–15077.
Optical activity and optical anisotropy in photomechanical crystals of chiral salicylidenephenylethylaminesAkifumi Takanabe, Masahito Tanaka, Kohei Johmoto, Hidehiro Uekusa, Tadashi Mori, Hideko Koshima*, and Toru Asahi*J. Am. Chem. Soc., 2016, 138, 15066–15077.Introducing chirality into photomechanical crystals is beneficial for the diversification of mechanical motion. Measurement of the chiroptical and optical anisotropic properties of chiral crystals is indispensable for evaluating photomechanical crystals. The platelike crystals of S- and R-enantiomers of photochromic N-3,5-di-tert-butylsalicylidene-1-phenylethylamine in enol form (enol-(S)-1 and enol-(R)-1) caused bending motion with twisting upon ultraviolet (UV) light irradiation, due to shrinkage along the length and width directions of the irradiated surface, based on the optimized crystal structure of the photoisomerized trans-keto-(S)-1. By employing the generalized high-accuracy universal polarimeter (G-HAUP), optical anisotropic (linear birefringence, LB; linear dichroism, LD) as well as chiroptical (circular birefringence, CB; circular dichroism, CD) spectra of both the enantiomeric crystals on the (001) face were simultaneously measured before and under continuous UV irradiation. The LD peak was observed at 330 nm in the negative sign, derived from the π–π* transition of the intramolecularly hydrogen-bonded salicylidenimino moiety. The CD spectra of the S and R crystals revealed the negative and positive Cotton effect at 330 nm, respectively, and new peaks appeared at 460 nm under UV light irradiation due to photoisomerization to the S and Rtrans-keto isomers at around 10% conversion. The CB and CD spectra evaluated by the HAUP measurement were opposite to those measured in the hexane solution, as well as those simulated by quantum chemical calculation. The dissymmetry parameter, g, of the enol-(S)-1 crystal along the c axis (0.013) was approximately 10 times larger than the g values in the solution (0.0010) and by calculation (0.0016).
@article{takanabe2016optical, title = {Optical activity and optical anisotropy in photomechanical crystals of chiral salicylidenephenylethylamines}, author = {Takanabe, Akifumi and Tanaka, Masahito and Johmoto, Kohei and Uekusa, Hidehiro and Mori, Tadashi and Koshima, Hideko and Asahi, Toru}, journal = {J. Am. Chem. Soc.}, volume = {138}, issue = {45}, pages = {15066--15077}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/jacs.6b09633}, url = {https://doi.org/10.1021/jacs.6b09633}, dimensions = {true}, tab = {paper}, } -
 Electrostatically promoted dynamic hybridization of glucans with cationic polythiopheneGaku Fukuhara*, Mami Imai, Denis Fuentealba, Yuki Ishida, Hiroki Kurohara, Cheng Yang, Tadashi Mori, Hiroshi Uyama, Cornelia Bohne, and Yoshihisa InoueOrg. Biomol. Chem., 2016, 14, 9741–9750.
Electrostatically promoted dynamic hybridization of glucans with cationic polythiopheneGaku Fukuhara*, Mami Imai, Denis Fuentealba, Yuki Ishida, Hiroki Kurohara, Cheng Yang, Tadashi Mori, Hiroshi Uyama, Cornelia Bohne, and Yoshihisa InoueOrg. Biomol. Chem., 2016, 14, 9741–9750.Hybridizing natural macromolecules with synthetic polymers is an efficient general method for constructing sophisticated supramolecular architectures. To comprehensively elucidate the controversial hybridization mechanism of glucans with synthetic polymers, the hybridization behaviors of triple-stranded curdlan (Cur) and schizophyllan (SPG) with cationic polythiophene (PyPT) were investigated in aqueous DMSO solutions by using UV-vis, circular dichroism (CD), fluorescence, fluorescence excitation, and NMR spectroscopy methods, as well as theoretical calculations, dynamic light scattering, and zeta potential measurements. Upon mixing with glucan, a hetero-triplex formed, which was dynamic and greatly accelerated by heating and by adding a base or a salt. The hetero-triplex disassembled into a hetero-duplex in highly basic solutions. Thus, polycationic polymers, such as PyPT, are expected to serve as a versatile tool for unzipping glucan homo-triplexes and promoting subsequent hybridization in aqueous solution, while the detailed mechanism elucidated in the present study contributes to the rational design of hybridization partners.
@article{fukuhara2016electrostatically, title = {Electrostatically promoted dynamic hybridization of glucans with cationic polythiophene}, author = {Fukuhara, Gaku and Imai, Mami and Fuentealba, Denis and Ishida, Yuki and Kurohara, Hiroki and Yang, Cheng and Mori, Tadashi and Uyama, Hiroshi and Bohne, Cornelia and Inoue, Yoshihisa}, journal = {Org. Biomol. Chem.}, volume = {14}, number = {41}, pages = {9741--9750}, year = {2016}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c6ob01353h}, url = {https://doi.org/10.1039/c6ob01353h}, dimensions = {true}, tab = {paper}, } -
 Supramolecular photochirogenesis with a higher-order complex: highly accelerated exclusively head-to-head photocyclodimerization of 2-anthracenecarboxylic acid via 2: 2 complexation with prolinolYuko Kawanami, Shin-ya Katsumata, Masaki Nishijima, Gaku Fukuhara, Kaori Asano, Takeyuki Suzuki, Cheng Yang, Asao Nakamura, Tadashi Mori, and Yoshihisa Inoue*J. Am. Chem. Soc., 2016, 138, 12187–12201.
Supramolecular photochirogenesis with a higher-order complex: highly accelerated exclusively head-to-head photocyclodimerization of 2-anthracenecarboxylic acid via 2: 2 complexation with prolinolYuko Kawanami, Shin-ya Katsumata, Masaki Nishijima, Gaku Fukuhara, Kaori Asano, Takeyuki Suzuki, Cheng Yang, Asao Nakamura, Tadashi Mori, and Yoshihisa Inoue*J. Am. Chem. Soc., 2016, 138, 12187–12201.An unprecedented 2:2 complex was shown to intervene in the enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid (A) mediated by a hydrogen-bonding template l-prolinol (P) to accelerate the formation of chiral anti-head-to-head and achiral syn-head-to-head cyclodimers in >99% combined yield with enhanced enantioselectivities of up to 72% ee for the former. The supramolecular complexation and photochirogenic behaviors, as well as the plausible structures, of intervening Am·Pn complexes (m, n = 1 or 2) were elucidated by combined theoretical and experimental spectroscopic, photophysical, and photochemical studies. Furthermore, the photochemical chiral amplification was achieved for the first time by utilizing the preferential 2:2 complexation of A with homochiral P to give normalized product enantioselectivities higher than those of the template used. The present strategy based on the higher-order hydrogen-bonding motif, which is potentially applicable to a variety of carboxylic acids and β-aminoalcohols, is not only conceptually new and expandable to other (photo)chirogenic and sensing systems but also may serve as a versatile tool for achieving photochemical asymmetric amplification and constructing chiral functional supramolecular architectures.
@article{kawanami2016supramolecular, title = {Supramolecular photochirogenesis with a higher-order complex: highly accelerated exclusively head-to-head photocyclodimerization of 2-anthracenecarboxylic acid via 2: 2 complexation with prolinol}, author = {Kawanami, Yuko and Katsumata, Shin-ya and Nishijima, Masaki and Fukuhara, Gaku and Asano, Kaori and Suzuki, Takeyuki and Yang, Cheng and Nakamura, Asao and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {138}, number = {37}, pages = {12187--12201}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/jacs.6b05598}, url = {https://doi.org/10.1021/jacs.6b05598}, dimensions = {true}, tab = {paper}, } -
 Combined Experimental and Theoretical Investigations on Optical Activities of Möbius Aromatic and Möbius Antiaromatic Hexaphyrin Phosphorus ComplexesTadashi Mori*, Takayuki Tanaka, Tomohiro Higashino, Kota Yoshida, and Atsuhiro OsukaJ. Phys. Chem. A, 2016, 120, 4241–4248.
Combined Experimental and Theoretical Investigations on Optical Activities of Möbius Aromatic and Möbius Antiaromatic Hexaphyrin Phosphorus ComplexesTadashi Mori*, Takayuki Tanaka, Tomohiro Higashino, Kota Yoshida, and Atsuhiro OsukaJ. Phys. Chem. A, 2016, 120, 4241–4248.Intrinsically chiral Möbius aromatic [28]hexaphyrin monophosphorus(V) and Möbius antiaromatic [30]hexaphyrin bisphosphorus(V) complexes have been optically resolved and their absolute configurations (ACs) were determined by combined experimental and theoretical investigations on their circular dichroisms (CDs). First elutes in chiral HPLC exhibited strong positive Cotton effects (CEs) at the B-band, characteristic for the ML configurations in their Möbius strips. Weak CEs at the Q-band, if attainable, complemented their AC assignment. The whole CD pattern and intensity were well reproduced by time-dependent approximate coupled cluster theory using model systems that omit five outward meso-aryl substituents (inward-meso-retained model), providing a solid basis for AC assignment. The cost efficient TD-DFT method with appropriate functionals for fully substituted (nontruncated) complexes well reproduced CEs around the B-band (but less satisfactory at the Q-band), also allows the rapid AC estimation for their Möbius strips. Observed difference in CDs between aromatic and antiaromatic hexaphyrins were better interpreted by their shifts in energy levels and altered interactions of relevant molecular orbitals, rather than small differences in Möbius geometries nor aromatic/antiaromatic character, despite the correlations recently claimed in planar π-systems.
@article{mori2016combined, title = {Combined Experimental and Theoretical Investigations on Optical Activities of Möbius Aromatic and Möbius Antiaromatic Hexaphyrin Phosphorus Complexes}, author = {Mori, Tadashi and Tanaka, Takayuki and Higashino, Tomohiro and Yoshida, Kota and Osuka, Atsuhiro}, journal = {J. Phys. Chem. A}, volume = {120}, number = {24}, pages = {4241--4248}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.6b03978}, url = {https://doi.org/10.1021/acs.jpca.6b03978}, dimensions = {true}, tab = {paper}, } -
 Inherently chiral azonia [6] helicene-modified β-cyclodextrin: Synthesis, characterization, and chirality sensing of underivatized amino acids in waterQinfei Huang, Liangwei Jiang, Wenting Liang, Jianchang Gui, Dingguo Xu, Wanhua Wu, Yoshito Nakai, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cheng Yang*J. Org. Chem., 2016, 81, 3430–3434.
Inherently chiral azonia [6] helicene-modified β-cyclodextrin: Synthesis, characterization, and chirality sensing of underivatized amino acids in waterQinfei Huang, Liangwei Jiang, Wenting Liang, Jianchang Gui, Dingguo Xu, Wanhua Wu, Yoshito Nakai, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cheng Yang*J. Org. Chem., 2016, 81, 3430–3434.The (P)- and (M)-3-azonia[6]helicenyl β-cyclodextrins exhibit l/d selectivities of up to 12.4 and P/M preferences of up to 28.2 upon complexation with underivatized proteinogenic amino acids in aqueous solution at pH 7.3.
@article{huang2016inherently, title = {Inherently chiral azonia [6] helicene-modified $\beta$-cyclodextrin: Synthesis, characterization, and chirality sensing of underivatized amino acids in water}, author = {Huang, Qinfei and Jiang, Liangwei and Liang, Wenting and Gui, Jianchang and Xu, Dingguo and Wu, Wanhua and Nakai, Yoshito and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa and Yang, Cheng}, journal = {J. Org. Chem.}, volume = {81}, number = {8}, pages = {3430--3434}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/acs.joc.6b00130}, url = {https://doi.org/10.1021/acs.joc.6b00130}, dimensions = {true}, tab = {paper}, } -
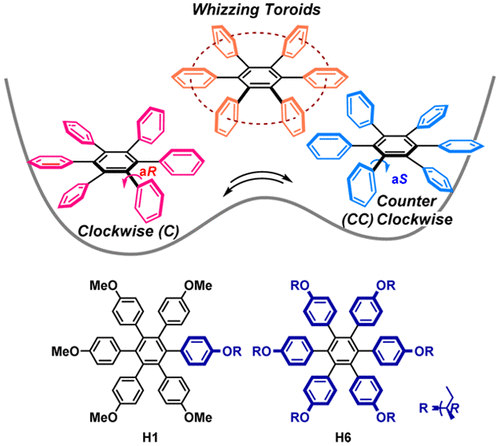 Toroidal interaction and propeller chirality of hexaarylbenzenes. Dynamic domino inversion revealed by combined experimental and theoretical circular dichroism studiesTomoyo Kosaka, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. Lett., 2016, 7, 783–788.
Toroidal interaction and propeller chirality of hexaarylbenzenes. Dynamic domino inversion revealed by combined experimental and theoretical circular dichroism studiesTomoyo Kosaka, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. Lett., 2016, 7, 783–788.Hexaarylbenzenes (HABs) have greatly attracted much attention due to their unique propeller-shaped structure and potential application in materials science, such as liquid crystals, molecular capsules/rotors, redox materials, nonlinear optical materials, as well as molecular wires. Less attention has however been paid to their propeller chirality. By introducing small point-chiral group(s) at the periphery of HABs, propeller chirality was effectively induced, provoking strong Cotton effects in the circular dichroism (CD) spectrum. Temperature and solvent polarity manipulate the dynamics of propeller inversion in solution. As such, whizzing toroids become more substantial in polar solvents and at an elevated temperature, where radial aromatic rings (propeller blades) prefer orthogonal alignment against the central benzene ring (C6 core), maximizing toroidal interactions.
@article{kosaka2016toroidal, title = {Toroidal interaction and propeller chirality of hexaarylbenzenes. Dynamic domino inversion revealed by combined experimental and theoretical circular dichroism studies}, author = {Kosaka, Tomoyo and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Phys. Chem. Lett.}, volume = {7}, number = {5}, pages = {783--788}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/acs.jpclett.6b00179}, url = {https://doi.org/10.1021/acs.jpclett.6b00179}, dimensions = {true}, tab = {paper}, } -
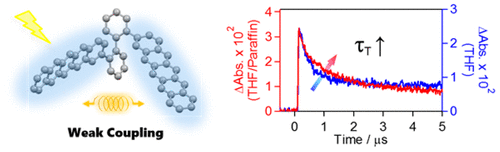 Long-lived triplet excited states of bent-shaped pentacene dimers by intramolecular singlet fissionTakao Sakuma, Hayato Sakai, Yasuyuki Araki, Tadashi Mori, Takehiko Wada, Nikolai V Tkachenko*, and Taku Hasobe*J. Phys. Chem. A, 2016, 120, 1867–1875.
Long-lived triplet excited states of bent-shaped pentacene dimers by intramolecular singlet fissionTakao Sakuma, Hayato Sakai, Yasuyuki Araki, Tadashi Mori, Takehiko Wada, Nikolai V Tkachenko*, and Taku Hasobe*J. Phys. Chem. A, 2016, 120, 1867–1875.Intramolecular singlet fission (ISF) is a promising photophysical process to construct more efficient light energy conversion systems as one excited singlet state converts into two excited triplet states. Herein we synthesized and evaluated bent-shaped pentacene dimers as a prototype of ISF to reveal intrinsic characters of triplet states (e.g., lifetimes of triplet excited states). In this study, meta-phenylene-bridged TIPS-pentacene dimer (PcD-3Ph) and 2,2′-bipheynyl bridged TIPS-pentacene dimer (PcD-Biph) were newly synthesized as bent-shaped dimers. In the steady-state spectroscopy, absorption and emission bands of these dimers were fully characterized, suggesting the appropriate degree of electronic coupling between pentacene moieties in these dimers. In addition, the electrochemical measurements were also performed to check the electronic interaction between two pentacene moieties. Whereas the successive two oxidation peaks owing to the delocalization were observed in a directly linked-pentacene dimer (PcD) by a single bond, the cyclic voltammograms in PcD-Biph and PcD-3Ph implied the weaker interaction compared to that of p-phenylene-bridged TIPS-pentacene dimer (PcD-4Ph) and PcD. The femtosecond and nanosecond transient absorption spectra clearly revealed the slower ISF process in bent-shaped pentacene dimers (PcD-Biph and PcD-3Ph), more notably, the slower relaxation of the excited triplet states in PcD-Biph and PcD-3Ph. Namely, the quantum yields of triplet states (ΦT) by ISF approximately remain constant (ca. 180–200%) in all dimer systems, whereas the lifetimes of the triplet excited states became much longer (up to 360 ns) in PcD-Biph as compared to PcD-4Ph (15 ns). Additionally, the lifetimes of the corresponding triplet states in PcD-Biph and PcD-3Ph were sufficiently affected by solvent viscosity. In particular, the lifetimes of PcD-Biph triplet state in THF/paraffin (1.0 μs) increased up to approximately three times as compared to that in THF (360 ns), whereas those of PcD-4Ph were quite similar in both solvent.
@article{sakuma2016long, title = {Long-lived triplet excited states of bent-shaped pentacene dimers by intramolecular singlet fission}, author = {Sakuma, Takao and Sakai, Hayato and Araki, Yasuyuki and Mori, Tadashi and Wada, Takehiko and Tkachenko, Nikolai V and Hasobe, Taku}, journal = {J. Phys. Chem. A}, volume = {120}, number = {11}, pages = {1867--1875}, year = {2016}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.6b00988}, url = {https://doi.org/10.1021/acs.jpca.6b00988}, dimensions = {true}, tab = {paper}, } -
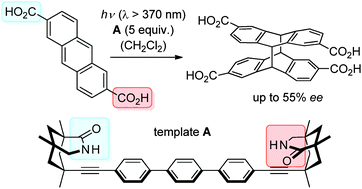 Enantioselective [4+4] photodimerization of anthracene-2,6-dicarboxylic acid mediated by a C2-symmetric chiral templateMark M Maturi, Gaku Fukuhara, Koichiro Tanaka, Yuko Kawanami, Tadashi Mori, Yoshihisa Inoue*, and Thorsten BachChem. Commun., 2016, 52, 1032–1035.
Enantioselective [4+4] photodimerization of anthracene-2,6-dicarboxylic acid mediated by a C2-symmetric chiral templateMark M Maturi, Gaku Fukuhara, Koichiro Tanaka, Yuko Kawanami, Tadashi Mori, Yoshihisa Inoue*, and Thorsten BachChem. Commun., 2016, 52, 1032–1035.A chiral template was constructed from 7-ethynyl-1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one by Sonogashira cross-coupling with 4,4′′-diiodoterphenyl and was shown to bind the title compound strongly by hydrogen bonding resulting in enantioselectivities of up to 55% enantiomeric excess (ee) in the [4+4] anthracene photodimerization.
@article{maturi2016enantioselective, title = {Enantioselective [4+4] photodimerization of anthracene-2,6-dicarboxylic acid mediated by a C2-symmetric chiral template}, author = {Maturi, Mark M and Fukuhara, Gaku and Tanaka, Koichiro and Kawanami, Yuko and Mori, Tadashi and Inoue, Yoshihisa and Bach, Thorsten}, journal = {Chem. Commun.}, volume = {52}, number = {5}, pages = {1032--1035}, year = {2016}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c5cc09107a}, url = {https://doi.org/10.1039/c5cc09107a}, dimensions = {true}, tab = {paper}, } -
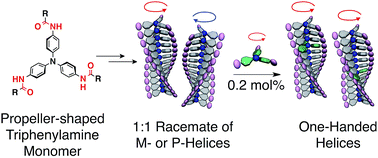 Dynamic propeller conformation for the unprecedentedly high degree of chiral amplification of supramolecular helicesTaehoon Kim, Tadashi Mori, Takuzo Aida, and Daigo Miyajima*Chem. Sci., 2016, 7, 6689–6694.
Dynamic propeller conformation for the unprecedentedly high degree of chiral amplification of supramolecular helicesTaehoon Kim, Tadashi Mori, Takuzo Aida, and Daigo Miyajima*Chem. Sci., 2016, 7, 6689–6694.An unprecedentedly high degree of chiral amplification of supramolecular helices in a sergeants and soldiers system was realized using a propeller-shaped molecule, triphenylamine (TPA), as the monomer. One sergeant controlled the handedness of 500 soldiers in supramolecular helices. We further demonstrated that a TPA derivative could switch its role from sergeant to soldier and vice versa depending on its partners. These achievements could be realized using the dynamic propeller conformation of TPA and provide new insights into supramolecular assemblies and the supramolecular chiral amplification of helices.
@article{kim2016dynamic, title = {Dynamic propeller conformation for the unprecedentedly high degree of chiral amplification of supramolecular helices}, author = {Kim, Taehoon and Mori, Tadashi and Aida, Takuzo and Miyajima, Daigo}, journal = {Chem. Sci.}, volume = {7}, number = {11}, pages = {6689--6694}, year = {2016}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c6sc02814d}, url = {https://doi.org/10.1039/c6sc02814d}, dimensions = {true}, tab = {paper}, }
2015
-
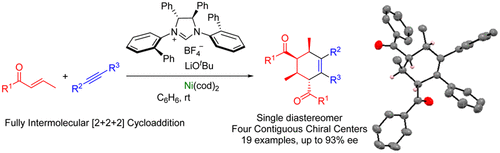 Nickel (0)/N-heterocyclic carbene-catalyzed asymmetric [2+ 2+ 2] cycloaddition of two enones and an alkyne: Access to cyclohexenes with four contiguous stereogenic centersRavindra Kumar, Hiromu Tokura, Akira Nishimura, Tadashi Mori, Yoichi Hoshimoto, Masato Ohashi, and Sensuke Ogoshi*Org. Lett., 2015, 17, 6018–6021.
Nickel (0)/N-heterocyclic carbene-catalyzed asymmetric [2+ 2+ 2] cycloaddition of two enones and an alkyne: Access to cyclohexenes with four contiguous stereogenic centersRavindra Kumar, Hiromu Tokura, Akira Nishimura, Tadashi Mori, Yoichi Hoshimoto, Masato Ohashi, and Sensuke Ogoshi*Org. Lett., 2015, 17, 6018–6021.A nickel(0)/chiral N-heterocyclic carbene (NHC)-catalyzed fully intermolecular, enantioselective [2 + 2 + 2] cycloaddition of two enones and an alkyne has been developed to access enantioenriched cyclohexenes. A single diastereomer was obtained with a successive generation of four contiguous stereogenic centers. The absolute configuration of cyclohexene derivative 3aa was determined by X-ray diffraction and circular dichroism (CD) spectral studies.
@article{kumar2015nickel, title = {Nickel (0)/N-heterocyclic carbene-catalyzed asymmetric [2+ 2+ 2] cycloaddition of two enones and an alkyne: Access to cyclohexenes with four contiguous stereogenic centers}, author = {Kumar, Ravindra and Tokura, Hiromu and Nishimura, Akira and Mori, Tadashi and Hoshimoto, Yoichi and Ohashi, Masato and Ogoshi, Sensuke}, journal = {Org. Lett.}, volume = {17}, number = {24}, pages = {6018--6021}, year = {2015}, publisher = {ACS Publications}, doi = {10.1021/acs.orglett.5b02983}, url = {https://doi.org/10.1021/acs.orglett.5b02983}, dimensions = {true}, tab = {paper}, } -
 Excited-state dynamics achieved ultimate stereocontrol of photocyclodimerization of anthracenecarboxylates on a glucose scaffoldGaku Fukuhara*, Kazuhiro Iida, Yuko Kawanami, Hidekazu Tanaka, Tadashi Mori, and Yoshihisa Inoue*J. Am. Chem. Soc., 2015, 137, 15007–15014.
Excited-state dynamics achieved ultimate stereocontrol of photocyclodimerization of anthracenecarboxylates on a glucose scaffoldGaku Fukuhara*, Kazuhiro Iida, Yuko Kawanami, Hidekazu Tanaka, Tadashi Mori, and Yoshihisa Inoue*J. Am. Chem. Soc., 2015, 137, 15007–15014.Near-perfect stereoselectivity was attained in the diastereodifferentiating [4 + 4] photocyclodimerization of 2-anthracenecarboxylates tethered to a glucose scaffold not by thermodynamically tuning the conformer equilibrium in the ground state but by kinetically controlling the conformer dynamics and reactivity in the excited state, which enabled us, after removal of the scaffold, to obtain a single enantiomer of chiral anti-head-to-head-cyclodimer in >99% optical and 96% chemical yield from an ensemble of four precursor conformers.
@article{fukuhara2015excited, title = {Excited-state dynamics achieved ultimate stereocontrol of photocyclodimerization of anthracenecarboxylates on a glucose scaffold}, author = {Fukuhara, Gaku and Iida, Kazuhiro and Kawanami, Yuko and Tanaka, Hidekazu and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {137}, number = {47}, pages = {15007--15014}, year = {2015}, publisher = {ACS Publications}, doi = {10.1021/jacs.5b09775}, url = {https://doi.org/10.1021/jacs.5b09775}, dimensions = {true}, tab = {paper}, } -
 pH-independent charge resonance mechanism for UV protective functions of shinorine and related mycosporine-like amino acidsKeisuke Matsuyama, Jun Matsumoto, Shogo Yamamoto, Keisuke Nagasaki, Yoshihisa Inoue, Masaki Nishijima, and Tadashi Mori*J. Phys. Chem. A, 2015, 119, 12722–12729.
pH-independent charge resonance mechanism for UV protective functions of shinorine and related mycosporine-like amino acidsKeisuke Matsuyama, Jun Matsumoto, Shogo Yamamoto, Keisuke Nagasaki, Yoshihisa Inoue, Masaki Nishijima, and Tadashi Mori*J. Phys. Chem. A, 2015, 119, 12722–12729.The UV-protective ability of mycosporine-like amino acids (MAAs) has been well documented and is believed to serve as a protecting agent for marine organisms from solar radiation. However, the effective UV absorption by MAAs has not been well correlated to MAA (neutral) structures. In this study, the origin of UV-protecting ability of MAAs was elucidated by experimental and theoretical spectroscopic investigations. The absorption maxima of mycosporine–glycine and shinorine in the UVA region were practically unaffected over a wide range of pH 4–10 and only slightly blue-shifted at pH 1–2. It was revealed that the zwitterionic nature of the amino acid residue facilitates the protonation to the chromophoric 3-aminocyclohexenone and 1-amino-3-iminocyclohexene moieties and the operation of the charge resonance in the protonated species well accounts for their allowed low-energy transitions in the UVA region. The RI-CC2/TZVP calculations on model systems in their protonated forms well reproduced the observed transition energies and oscillator strengths of MAAs, only with insignificant systematic overestimations of the both values. The slight hypsochromic shifts at pH 1–2 were explained by (partial) protonation to a carboxylate anion in the amino acid residue, as confirmed by theory. Fluorescence spectral investigations of shinorine were also performed for the first time in water to confirm the effective nonradiative deactivation. Consequently, this study unequivocally demonstrated that the 3-aminocyclohexenone as well as 1-amino-3-iminocyclohexene moieties, which are readily protonated at a wide range of pH, are responsible for the UV-protective ability of aqueous solution of MAAs.
@article{matsuyama2015ph, title = {pH-independent charge resonance mechanism for UV protective functions of shinorine and related mycosporine-like amino acids}, author = {Matsuyama, Keisuke and Matsumoto, Jun and Yamamoto, Shogo and Nagasaki, Keisuke and Inoue, Yoshihisa and Nishijima, Masaki and Mori, Tadashi}, journal = {J. Phys. Chem. A}, volume = {119}, number = {51}, pages = {12722--12729}, year = {2015}, publisher = {ACS Publications}, doi = {10.1021/acs.jpca.5b09988}, url = {https://doi.org/10.1021/acs.jpca.5b09988}, dimensions = {true}, tab = {paper}, } -
 Helix sense-selective supramolecular polymerization seeded by a one-handed helical polymeric assemblyWei Zhang, Wusong Jin, Takanori Fukushima*, Tadashi Mori, and Takuzo Aida*. Am. Chem. Soc., 2015, 137, 13792–13795.
Helix sense-selective supramolecular polymerization seeded by a one-handed helical polymeric assemblyWei Zhang, Wusong Jin, Takanori Fukushima*, Tadashi Mori, and Takuzo Aida*. Am. Chem. Soc., 2015, 137, 13792–13795.Helix sense-selective supramolecular polymerization was achieved using a one-handed helical nanotubular polymeric assembly as a seed. First, bipyridine (BPY)-appended achiral hexabenzocoronene (BPYHBC) was copolymerized noncovalently with chiral BPYHBCS (or BPYHBCR) at a molar ratio of 9:1, which, via the sergeants-and-soldiers effect, afforded a P-helical (or M-helical) nanotube, which was then treated with Cu2+ to transform into structurally robust (BPY)CuNT(P) (or (BPY)CuNT(M)) with a Cu2+/BPY coordination polymer shell. Helical seeds (BPY)CuNT(P) and (BPY)CuNT(M) brought about the controlled assembly of fluorinated chiral FHBCS and FHBCR as well as achiral FHBC to yield one-handed helical nanotubular supramolecular block copolymers, in which the helical senses of the newly formed block segments were solely determined by those of the helical seeds employed. Noteworthy, FHBCS and FHBCR alone without the helical seeds form ill-defined agglomerates. Attempted supramolecular polymerization of a racemic mixture of FHBCS and FHBCR from (BPY)CuNT(P) (or (BPY)CuNT(M)) resulted in its chiral separation, affording P-helical (or M-helical) diastereomeric block segments composed of FHBCS and FHBCR with different thermodynamic properties.
@article{zhang2015helix, title = {Helix sense-selective supramolecular polymerization seeded by a one-handed helical polymeric assembly}, author = {Zhang, Wei and Jin, Wusong and Fukushima, Takanori and Mori, Tadashi and Aida, Takuzo}, journal = {. Am. Chem. Soc.}, volume = {137}, number = {43}, pages = {13792--13795}, year = {2015}, publisher = {ACS Publications}, doi = {10.1021/jacs.5b09878}, url = {https://doi.org/10.1021/jacs.5b09878}, dimensions = {true}, tab = {paper}, } -
 Metal–Organic Nanotube with Helical and Propeller-Chiral Motifs Composed of a C 10-Symmetric Double-Decker NanoringHiroshi Yamagishi, Takahiro Fukino*, Daisuke Hashizume, Tadashi Mori, Yoshihisa Inoue, Takaaki Hikima, Masaki Takata, and Takuzo Aida*J. Am. Chem. Soc., 2015, 137, 7628–7631.
Metal–Organic Nanotube with Helical and Propeller-Chiral Motifs Composed of a C 10-Symmetric Double-Decker NanoringHiroshi Yamagishi, Takahiro Fukino*, Daisuke Hashizume, Tadashi Mori, Yoshihisa Inoue, Takaaki Hikima, Masaki Takata, and Takuzo Aida*J. Am. Chem. Soc., 2015, 137, 7628–7631.Coassembly of an achiral ferrocene-cored tetratopic pyridyl ligand (FcL) with AgBF4 in CH2Cl2/MeCN (7:3 v/v) containing chiral Bu4N+ (+)- or (−)-menthylsulfate (MS*–) results in the formation of an “optically active” metal–organic nanotube (FcNT) composed of a C10-symmetric double-decker nanoring featuring 10 FcL units and 20 Ag+ ions. The circular dichroism spectrum of FcNT along with its 2D X-ray diffraction (2D XRD) pattern indicates that the constituent metal–organic nanorings in FcNT stack one-handed helically on top of each other. A crystal structure of the dimeric double-decker model complex (Ag2(FcL′)2) from a ditopic ferrocene ligand (FcL′) and AgBF4 allowed for confirming the binding of MS*– onto the Ag+ center of the complex. The results of detailed spectroscopic studies indicate that in its double-decker aromatic arrays, FcNT possibly possesses propeller-chiral twists in addition to the helically chiral structure, where the former is considerably more dynamic than the latter. Notably, both chiral structural motifs responded nonlinearly to an enantiomeric excess of MS*– (majority rule) though with no stereochemical influence on one another.
@article{yamagishi2015metal, title = {Metal--Organic Nanotube with Helical and Propeller-Chiral Motifs Composed of a C 10-Symmetric Double-Decker Nanoring}, author = {Yamagishi, Hiroshi and Fukino, Takahiro and Hashizume, Daisuke and Mori, Tadashi and Inoue, Yoshihisa and Hikima, Takaaki and Takata, Masaki and Aida, Takuzo}, journal = {J. Am. Chem. Soc.}, volume = {137}, number = {24}, pages = {7628--7631}, year = {2015}, publisher = {ACS Publications}, doi = {10.1021/jacs.5b04386}, url = {https://doi.org/10.1021/jacs.5b04386}, dimensions = {true}, tab = {paper}, } -
 A rational strategy for the realization of chain-growth supramolecular polymerizationJiheong Kang, Daigo Miyajima*, Tadashi Mori, Yoshihisa Inoue, Yoshimitsu Itoh, and Takuzo Aida*Science, 2015, 347, 646–651.
A rational strategy for the realization of chain-growth supramolecular polymerizationJiheong Kang, Daigo Miyajima*, Tadashi Mori, Yoshihisa Inoue, Yoshimitsu Itoh, and Takuzo Aida*Science, 2015, 347, 646–651.Over the past decade, major progress in supramolecular polymerization has had a substantial effect on the design of functional soft materials. However, despite recent advances, most studies are still based on a preconceived notion that supramolecular polymerization follows a step-growth mechanism, which precludes control over chain length, sequence, and stereochemical structure. Here we report the realization of chain-growth polymerization by designing metastable monomers with a shape-promoted intramolecular hydrogen-bonding network. The monomers are conformationally restricted from spontaneous polymerization at ambient temperatures but begin to polymerize with characteristics typical of a living mechanism upon mixing with tailored initiators. The chain growth occurs stereoselectively and therefore enables optical resolution of a racemic monomer.
@article{kang2015rational, title = {A rational strategy for the realization of chain-growth supramolecular polymerization}, author = {Kang, Jiheong and Miyajima, Daigo and Mori, Tadashi and Inoue, Yoshihisa and Itoh, Yoshimitsu and Aida, Takuzo}, journal = {Science}, volume = {347}, number = {6222}, pages = {646--651}, year = {2015}, publisher = {American Association for the Advancement of Science}, doi = {10.1126/science.aaa4249}, url = {https://doi.org/10.1126/science.aaa4249}, dimensions = {true}, tab = {paper}, } -
 Contrasting Behaviour of Exciplex Ensembles in the Diastereodifferentiating Paternò–Büchi Reaction of Chiral Cyanobenzoate with Naphthyl-and Phenylethenes on Direct or Charge-Transfer ExcitationKeisuke Nagasaki, Yoshihisa Inoue, and Tadashi Mori*Aust. J. Chem., 2015, 68, 1693–1699.
Contrasting Behaviour of Exciplex Ensembles in the Diastereodifferentiating Paternò–Büchi Reaction of Chiral Cyanobenzoate with Naphthyl-and Phenylethenes on Direct or Charge-Transfer ExcitationKeisuke Nagasaki, Yoshihisa Inoue, and Tadashi Mori*Aust. J. Chem., 2015, 68, 1693–1699.The diastereodifferentiating Paternò–Büchi reaction of chiral cyanobenzoate with 1-(1-naphthyl)-1-phenylethene was compared with those with 1,1-diphenylethene on direct and charge-transfer excitations. By desymmetrization of the donor, four diastereomeric oxetane products were formed on photolysis in excellent combined yields. Increase in donor strength induced a stronger charge-transfer interaction both in ground and excited states. Thus, the difference in diastereoselectivities with two different excitation modes (i.e. direct versus charge-transfer) became less significant with a naphthyl derivative as donor. A subtle change of donor–acceptor interaction was shown to have profound effect on the nature of the excited-state complexes and thus the product (stereo)selectivities. Despite a small temperature dependence, an Eyring-type study on the diastereoselectivities confirmed that the excited charge-transfer complex is an excited species distinct from the conventional exciplex.
@article{nagasaki2015contrasting, title = {Contrasting Behaviour of Exciplex Ensembles in the Diastereodifferentiating Paternò--Büchi Reaction of Chiral Cyanobenzoate with Naphthyl-and Phenylethenes on Direct or Charge-Transfer Excitation}, author = {Nagasaki, Keisuke and Inoue, Yoshihisa and Mori, Tadashi}, journal = {Aust. J. Chem.}, volume = {68}, number = {11}, pages = {1693--1699}, year = {2015}, publisher = {CSIRO Publishing}, doi = {10.1071/ch15404}, url = {https://doi.org/10.1071/ch15404}, dimensions = {true}, tab = {paper}, } - Studies on Circular Dichroisms and Circular Polarized Luminescence Properties Based on Helical Structure of Helicene DerivativesTadashi Mori*Toyota Kenkyu Hokoku, 2015, 68, 173–174.
@article{mori2015studies, title = {Studies on Circular Dichroisms and Circular Polarized Luminescence Properties Based on Helical Structure of Helicene Derivatives}, author = {Mori, Tadashi}, journal = {Toyota Kenkyu Hokoku}, volume = {68}, pages = {173--174}, year = {2015} }
2014
-
 Exciplex Ensemble Modulated by Excitation Mode in Intramolecular Charge-Transfer Dyad: Effects of Temperature, Solvent Polarity, and Wavelength on Photochemistry and Photophysics of Tethered Naphthalene–Dicyanoethene SystemYoshiaki Aoki, Nobuo Matsuki, Tadashi Mori*, Hiroshi Ikeda, and Yoshihisa Inoue*Org. Lett., 2014, 16, 4888–4891.
Exciplex Ensemble Modulated by Excitation Mode in Intramolecular Charge-Transfer Dyad: Effects of Temperature, Solvent Polarity, and Wavelength on Photochemistry and Photophysics of Tethered Naphthalene–Dicyanoethene SystemYoshiaki Aoki, Nobuo Matsuki, Tadashi Mori*, Hiroshi Ikeda, and Yoshihisa Inoue*Org. Lett., 2014, 16, 4888–4891.Solvent, temperature, and excitation wavelength significantly affected the photochemical outcomes of a naphthalene–dicyanoethene system tethered by different number (n) of methylene groups (1–3). The effect of irradiation wavelength was almost negligible for 2a but pronounced for 3a. The temperature dependence and theoretical calculations indicated the diversity of exciplex conformations, an ensemble of which can be effectively altered by changing excitation wavelength to eventually switch the regioselectivity of photoreactions.
@article{aoki2014exciplex, title = {Exciplex Ensemble Modulated by Excitation Mode in Intramolecular Charge-Transfer Dyad: Effects of Temperature, Solvent Polarity, and Wavelength on Photochemistry and Photophysics of Tethered Naphthalene--Dicyanoethene System}, author = {Aoki, Yoshiaki and Matsuki, Nobuo and Mori, Tadashi and Ikeda, Hiroshi and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {16}, number = {18}, pages = {4888--4891}, year = {2014}, publisher = {ACS Publications}, doi = {10.1021/ol502394g}, url = {https://doi.org/10.1021/ol502394g}, dimensions = {true}, tab = {paper}, } -
 C-5-Symmetric chiral corannulenes: desymmetrization of bowl inversion equilibrium via “intramolecular” hydrogen-bonding networkJiheong Kang, Daigo Miyajima*, Yoshimitsu Itoh, Tadashi Mori, Hiroki Tanaka, Masahito Yamauchi, Yoshihisa Inoue, Soichiro Harada, and Takuzo Aida*J. Am. Chem. Soc., 2014, 136, 10640–10644.
C-5-Symmetric chiral corannulenes: desymmetrization of bowl inversion equilibrium via “intramolecular” hydrogen-bonding networkJiheong Kang, Daigo Miyajima*, Yoshimitsu Itoh, Tadashi Mori, Hiroki Tanaka, Masahito Yamauchi, Yoshihisa Inoue, Soichiro Harada, and Takuzo Aida*J. Am. Chem. Soc., 2014, 136, 10640–10644.Because of a rapid conformational inversion, bowl-shaped C5-symmetric corannulenes, though geometrically chiral, have not been directly resolved into their enantiomers. However, if this inversion equilibrium can be desymmetrized, chiral corannulenes enriched in either enantiomer can be obtained. We demonstrated this possibility using pentasubstituted corannulenes 4 and 5 carrying amide-appended thioalkyl side chains. Compound 4 displays chiroptical activity in a chiral hydrocarbon such as limonene. Because compound 5 carries a chiral center in the side chains, its enantiomers 5R and 5S show chiroptical activity even in achiral solvents such as CHCl3 and methylcyclohexane. In sharp contrast, when the side chains bear no amide functionality (1 and 2R), no chiroptical activity emerges even in limonene or with a chiral center in the side chains. Detailed investigations revealed that the peripheral amide units in 4 and 5 are hydrogen-bonded only “intramolecularly” along the corannulene periphery, affording cyclic amide networks with clockwise and anticlockwise geometries. Although this networking gives rise to four stereoisomers, only two, which are enantiomeric to one another, are suggested computationally to exist in the equilibrated system. In a chiral environment (chiral solvent or side chain), their thermodynamic stabilities are certainly unequal, so the bowl-inversion equilibrium can be desymmetrized. However, this is not the case when the system contains a protic solvent that can deteriorate the hydrogen-bonding network. When the enantiomeric purity of limonene as the solvent is varied, the chiroptical activity of the corannulene core changes nonlinearly with its enantiomeric excess (majority rule).
@article{kang2014c, title = {C-5-Symmetric chiral corannulenes: desymmetrization of bowl inversion equilibrium via “intramolecular” hydrogen-bonding network}, author = {Kang, Jiheong and Miyajima, Daigo and Itoh, Yoshimitsu and Mori, Tadashi and Tanaka, Hiroki and Yamauchi, Masahito and Inoue, Yoshihisa and Harada, Soichiro and Aida, Takuzo}, journal = {J. Am. Chem. Soc.}, volume = {136}, number = {30}, pages = {10640--10644}, year = {2014}, publisher = {ACS Publications}, doi = {10.1021/ja505941b}, url = {https://doi.org/10.1021/ja505941b}, dimensions = {true}, tab = {paper}, } -
 ABC-Type meso-Triaryl-Substituted SubporphyrinsKota Yoshida, Hirotaka Mori, Takayuki Tanaka, Tadashi Mori*, and Atsuhiro Osuka*Eur. J. Org. Chem., 2014, 2014, 3997–4004.
ABC-Type meso-Triaryl-Substituted SubporphyrinsKota Yoshida, Hirotaka Mori, Takayuki Tanaka, Tadashi Mori*, and Atsuhiro Osuka*Eur. J. Org. Chem., 2014, 2014, 3997–4004.ABC-Type subporphyrins 5a–5h, which bear three different meso-aryl substituents, were rationally synthesized by condensation of AB-type tripyrranes and aroyl chlorides. ABC-Type subporphyrins 5i and 5j were synthesized by Pd-catalyzed amination reaction of 4-bromophenyl subporphyrins 5e and 5h, respectively. Comparative studies on these ABC-type subporphyrins with A3-type subporphyrins and A2B-type subporphyrins revealed that substituent effects are mostly simple superpositions of individual substituents but cooperative effects are recognized for subporphyrins which bear both electron-donating and electron-withdrawing substituents. Despite extensive attempts, enantiomeric separation of B-methoxy ABC-type subporphyrins was unsuccessful, whereas B-phenylated ABC-type subporphyrins were separated to pure enantiomers. Separated enantiomers showed weak but distinct CD signals reflecting the chiral clockwise/anticlockwise arrangements of the meso-aryl substituents. These results suggest facile racemization through SN1-type heterolysis of the B–OMe bond in solution.
@article{yoshida2014abc, title = {ABC-Type meso-Triaryl-Substituted Subporphyrins}, author = {Yoshida, Kota and Mori, Hirotaka and Tanaka, Takayuki and Mori, Tadashi and Osuka, Atsuhiro}, journal = {Eur. J. Org. Chem.}, volume = {2014}, number = {19}, pages = {3997--4004}, year = {2014}, publisher = {Wiley Online Library}, doi = {10.1002/ejoc.201402435}, url = {https://doi.org/10.1002/ejoc.201402435}, dimensions = {true}, tab = {paper}, } -
 Ammonia-Driven Chirality Inversion and Enhancement in Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Diguanidino-γ-cyclodextrinJiabin Yao, Zhiqiang Yan, Jiecheng Ji, Wanhua Wu, Cheng Yang*, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, and Yoshihisa Inoue*J. Am. Chem. Soc., 2014, 136, 6916–6919.
Ammonia-Driven Chirality Inversion and Enhancement in Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Diguanidino-γ-cyclodextrinJiabin Yao, Zhiqiang Yan, Jiecheng Ji, Wanhua Wu, Cheng Yang*, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, and Yoshihisa Inoue*J. Am. Chem. Soc., 2014, 136, 6916–6919.In the supramolecular photocyclodimerization of 2-anthracenecarboxylate mediated by 6A,6D-diguanidino-γ-cyclodextrin (CD), the chiral sense and enantiomeric excess of the photoproduct were dynamic functions of temperature and cosolvent to afford the (M)-anti head-to-head cyclodimer in 64 ee in aqueous methanol at −70 °C but the antipodal (P)-isomer in 86 ee in aqueous ammonia at −85 °C, while the corresponding diamino-γ-CD host did not show such unusual photochirogenic behaviors. The ee landscape was very steep against the temperature and sign-inverted against the ammonia content to reveal the opposite temperature dependence at low and high ammonia contents, for which an altered solvent structure and/or guanidinium–carboxylate interaction mode would be responsible.
@article{yao2014ammonia, title = {Ammonia-Driven Chirality Inversion and Enhancement in Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Diguanidino-γ-cyclodextrin}, author = {Yao, Jiabin and Yan, Zhiqiang and Ji, Jiecheng and Wu, Wanhua and Yang, Cheng and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {136}, number = {19}, pages = {6916--6919}, year = {2014}, publisher = {ACS Publications}, doi = {10.1021/ja5032908}, url = {https://doi.org/10.1021/ja5032908}, dimensions = {true}, tab = {paper}, } -
 Sign inversion of circularly polarized luminescence by geometry manipulation of four naphthalene units introduced into a tartaric acid scaffoldTomoyuki Amako, Kazuki Nakabayashi, Tadashi Mori*, Yoshihisa Inoue, Michiya Fujiki, and Yoshitane Imai*Chem. Commun., 2014, 50, 12836–12839.
Sign inversion of circularly polarized luminescence by geometry manipulation of four naphthalene units introduced into a tartaric acid scaffoldTomoyuki Amako, Kazuki Nakabayashi, Tadashi Mori*, Yoshihisa Inoue, Michiya Fujiki, and Yoshitane Imai*Chem. Commun., 2014, 50, 12836–12839.Tethering four 1- versus 2-naphthyls to the same chiral scaffold derived from tartaric acid led to oppositely signed circularly polarized luminescence (CPL) and circular dichroism (CD) in solution, which not only reveals the decisive role of the spatial arrangement of chromophores/fluorophores in determining the chiroptical behaviors but also provides us with a versatile tool for switching the signs of CPL and CD without using the antipodal scaffold.
@article{amako2014sign, title = {Sign inversion of circularly polarized luminescence by geometry manipulation of four naphthalene units introduced into a tartaric acid scaffold}, author = {Amako, Tomoyuki and Nakabayashi, Kazuki and Mori, Tadashi and Inoue, Yoshihisa and Fujiki, Michiya and Imai, Yoshitane}, journal = {Chem. Commun.}, volume = {50}, number = {85}, pages = {12836--12839}, year = {2014}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c4cc04228j}, url = {https://doi.org/10.1039/c4cc04228j}, dimensions = {true}, tab = {paper}, } -
 Mammalian serum albumins as a chiral mediator library for bio-supramolecular photochirogenesis: optimizing enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylateMasaki Nishijima*, Masato Goto, Mayu Fujikawa, Cheng Yang, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2014, 50, 14082–14085.
Mammalian serum albumins as a chiral mediator library for bio-supramolecular photochirogenesis: optimizing enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylateMasaki Nishijima*, Masato Goto, Mayu Fujikawa, Cheng Yang, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2014, 50, 14082–14085.A simple strategy for choosing optimal bio-supramolecular mediators from the mammalian serum albumin library is proposed for bimolecular photochirogenic reactions. Thus, the enantiodifferentiating photocyclodimerization of 2-anthracencecarboxylate (AC) was optimized in chemical and optical yields, when mediated by porcine and canine serum albumins, both of which bound two AC molecules in the first productive site to give the (P)-enantiomer of syn-head-to-tail-cyclodimer in 69% yield and 89% enantiomeric excess (ee) for the former but the (M)-enantiomer in 77% yield and 97% ee for the latter.
@article{nishijima2014mammalian, title = {Mammalian serum albumins as a chiral mediator library for bio-supramolecular photochirogenesis: optimizing enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate}, author = {Nishijima, Masaki and Goto, Masato and Fujikawa, Mayu and Yang, Cheng and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Commun.}, volume = {50}, number = {91}, pages = {14082--14085}, year = {2014}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c4cc04818k}, url = {https://doi.org/10.1039/c4cc04818k}, dimensions = {true}, tab = {paper}, } -
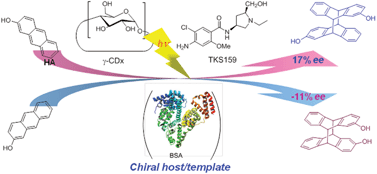 Supramolecular photocyclodimerization of 2-hydroxyanthracene with a chiral hydrogen-bonding template, cyclodextrin and serum albuminGaku Fukuhara, Hiroaki Umehara, Saki Higashino, Masaki Nishijima, Cheng Yang, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2014, 13, 162–171.
Supramolecular photocyclodimerization of 2-hydroxyanthracene with a chiral hydrogen-bonding template, cyclodextrin and serum albuminGaku Fukuhara, Hiroaki Umehara, Saki Higashino, Masaki Nishijima, Cheng Yang, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2014, 13, 162–171.2-Hydroxyanthracene (HA) in its neutral form smoothly photocyclodimerized to four stereoisomeric [4 + 4]-cyclodimers, which were isolated and characterized for the first time, whereas the anionic form of HA turned out to be photochemically inert. Enantiodifferentiating photocyclodimerization of HA in the presence of a chiral hydrogen-bonding template (TKS159), γ-cyclodextrin (γ-CDx) and bovine serum albumin (BSA) was examined to afford chiral syn-head-to-tail and anti-head-to-head cyclodimers in modest enantiomeric excesses with TKS159 and γ-CDx, but practically no photocyclodimerization proceeded in the presence of BSA probably due to the ionization of HA in the binding sites.
@article{fukuhara2014supramolecular, title = {Supramolecular photocyclodimerization of 2-hydroxyanthracene with a chiral hydrogen-bonding template, cyclodextrin and serum albumin}, author = {Fukuhara, Gaku and Umehara, Hiroaki and Higashino, Saki and Nishijima, Masaki and Yang, Cheng and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Photochem. Photobiol. Sci.}, volume = {13}, number = {2}, pages = {162--171}, year = {2014}, publisher = {Springer}, doi = {10.1039/c3pp50127b}, url = {https://doi.or10.1039/c3pp50127b}, dimensions = {true}, tab = {paper}, } -
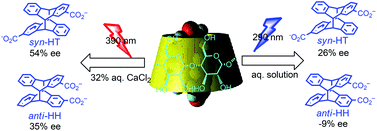 Manipulating γ-cyclodextrin-mediated photocyclodimerization of anthracenecarboxylate by wavelength, temperature, solvent and hostCheng Yang*, Qian Wang, Masahito Yamauchi, Jabing Yao, Dayang Zhou, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yu Liu*, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2014, 13, 190–198.
Manipulating γ-cyclodextrin-mediated photocyclodimerization of anthracenecarboxylate by wavelength, temperature, solvent and hostCheng Yang*, Qian Wang, Masahito Yamauchi, Jabing Yao, Dayang Zhou, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yu Liu*, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2014, 13, 190–198.Wavelength effects on the enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate (AC) mediated by native and modified γ-cyclodextrins (CDs) were examined in different solvents at varying temperatures to manipulate the photochirogenic outcomes beyond the thermodynamically determined re/si-enantiotopic face selectivity upon 2 : 1 complexation of AC with CD in the ground state. Indeed, the stereochemical outcomes, i.e. syn/anti, head-to-tail/head-to-head (HT/HH) and in particular enantiomer ratios, were critical functions of the irradiation wavelength, irrespective of the CD host employed. Furthermore, the wavelength effects observed strongly depended on the host structure, reaction temperature and solvent employed, for which altered stacking geometry of the complexed AC pair is thought to be responsible. By optimizing the irradiation wavelength, chiral host, temperature and solvent, an enantiomeric excess of up to 54 and −37% were achieved for chiral syn-HT and anti-HH dimers.
@article{yang2014manipulating, title = {Manipulating $\gamma$-cyclodextrin-mediated photocyclodimerization of anthracenecarboxylate by wavelength, temperature, solvent and host}, author = {Yang, Cheng and Wang, Qian and Yamauchi, Masahito and Yao, Jabing and Zhou, Dayang and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Liu, Yu and Inoue, Yoshihisa}, journal = {Photochem. Photobiol. Sci.}, volume = {13}, number = {2}, pages = {190--198}, year = {2014}, publisher = {Springer}, doi = {10.1039/c3pp50255d}, url = {https://doi.org/10.1039/c3pp50255d}, dimensions = {true}, tab = {paper}, } - Combined experimental and theoretical investigation of chiroptical properties of helicenes and related moleculesTadashi Mori*Symmetry, 2014, 25, 109–112.
@article{mori2014combined, title = {Combined experimental and theoretical investigation of chiroptical properties of helicenes and related molecules}, author = {Mori, Tadashi}, journal = {Symmetry}, volume = {25}, issue = {2}, pages = {109--112}, year = {2014}, publisher = {Symmetry: Culture and Science} }
2013
-
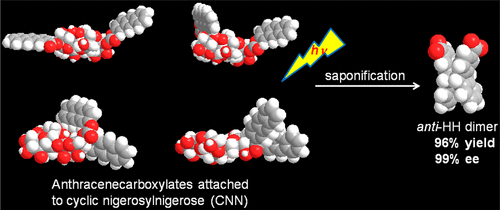 Diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylates tethered to a cyclic tetrasaccharide scaffold: Critical control of photoreactivity and stereoselectivityGaku Fukuhara*, Tomohiro Nakamura, Yuko Kawanami, Cheng Yang, Tadashi Mori, Hiroyuki Hiramatsu, Yasufumi Dan-Oh, Tomoyuki Nishimoto, Kazuo Tsujimoto, and Yoshihisa Inoue*J. Org. Chem., 2013, 78, 10996–11006.
Diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylates tethered to a cyclic tetrasaccharide scaffold: Critical control of photoreactivity and stereoselectivityGaku Fukuhara*, Tomohiro Nakamura, Yuko Kawanami, Cheng Yang, Tadashi Mori, Hiroyuki Hiramatsu, Yasufumi Dan-Oh, Tomoyuki Nishimoto, Kazuo Tsujimoto, and Yoshihisa Inoue*J. Org. Chem., 2013, 78, 10996–11006.From a complex mixture of mono- and di-2-anthracenecarboxylic acid (AC) esters of cyclic nigerosylnigerose (CNN), two monoesters (2B and 6A) and four diesters in which AC was introduced on the transannular B/D (2B2D), adjacent A/B and A/D (6A2B and 6A2D), and same B/B (2B3B) nigerose rings were isolated. Possessing two ACs at distant positions, 2B2D and 6A2D showed negative Cotton effects for the 1Bb band, the intensities of which were stronger than that of 6A. 2B2D and 6A2D slowly photocyclodimerized to give HH dimers 3* and 4 with 57% and 81% HH selectivity, respectively, which were appreciably higher than that for 6A (34%), while the enantiomeric excesses (ee’s) of anti-HH dimer 3* were 2% and −18%, respectively. In contrast, 6A2B and 2B3B carrying two ACs on adjacent A and B rings or at vicinal positions on the B ring, respectively, exhibited strong positive CD couplets, the amplitudes of which amounted to 97 and 409 M–1 cm–1, respectively. Upon irradiation, 6A2B afforded 3* with −62% ee and 4 in 96% combined yield, whereas 2B3B gave almost exclusively 3* with −99% ee in 96% yield, likely as a result of the introduction of two ACs at the vicinal positions of the rigid CNN scaffold.
@article{fukuhara2013diastereodifferentiating, title = {Diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylates tethered to a cyclic tetrasaccharide scaffold: Critical control of photoreactivity and stereoselectivity}, author = {Fukuhara, Gaku and Nakamura, Tomohiro and Kawanami, Yuko and Yang, Cheng and Mori, Tadashi and Hiramatsu, Hiroyuki and Dan-Oh, Yasufumi and Nishimoto, Tomoyuki and Tsujimoto, Kazuo and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {78}, number = {21}, pages = {10996--11006}, year = {2013}, publisher = {ACS Publications}, doi = {10.1021/jo401977f}, url = {https://doi.org/10.1021/jo401977f}, dimensions = {true}, tab = {paper}, } -
 Absolute configuration determination of the anti-head-to-head photocyclodimer of anthracene-2-carboxylic acid through cocrystallization with l-prolinolYuko Kawanami, Hidekazu Tanaka, Jun-ichi Mizoguchi, Nobuko Kanehisa, Gaku Fukuhara, Masaki Nishijima, Tadashi Mori, and Yoshihisa InoueActa Cryst. C, 2013, 69, 1411–1413.
Absolute configuration determination of the anti-head-to-head photocyclodimer of anthracene-2-carboxylic acid through cocrystallization with l-prolinolYuko Kawanami, Hidekazu Tanaka, Jun-ichi Mizoguchi, Nobuko Kanehisa, Gaku Fukuhara, Masaki Nishijima, Tadashi Mori, and Yoshihisa InoueActa Cryst. C, 2013, 69, 1411–1413.The absolute configuration has been established of the enantio pure anti-head-to-head cyclo dimer of anthracene-2-carboxylic acid (AC) cocrystallized with L-propinol and dichloromethane [systematic name: (S)-2-(hydroxymethyl)pyrrolidin-1-ium (5R,6S,11R,12S)-8-carboxy-5,6,11,12-tetrahydro-5,12:6,11-bis([1,2]benzeno)dibenzo[a,e][8]annulene-2-carboxylate dichloromethane monosolvate], C5H12NO+·C30H19O4-·CH2Cl2. In the crystal structure, the AC dimer interacts with L-prolinol through a nine-membered hydrogen-bonded ring [R22(9)], while the dichloromethane molecule is incorporated to fill the void space. The absolute configuration determined in this study verifies a recent assignment made by comparing theoretical versus experimental circular dichroism spectra.
@article{kawanami2013absolute, title = {Absolute configuration determination of the anti-head-to-head photocyclodimer of anthracene-2-carboxylic acid through cocrystallization with l-prolinol}, author = {Kawanami, Yuko and Tanaka, Hidekazu and Mizoguchi, Jun-ichi and Kanehisa, Nobuko and Fukuhara, Gaku and Nishijima, Masaki and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Acta Cryst. C}, volume = {69}, number = {11}, pages = {1411--1413}, year = {2013}, publisher = {International Union of Crystallography}, doi = {10.1107/S0108270113028461}, url = {https://doi.org/10.1107/S0108270113028461}, dimensions = {true}, tab = {paper}, } -
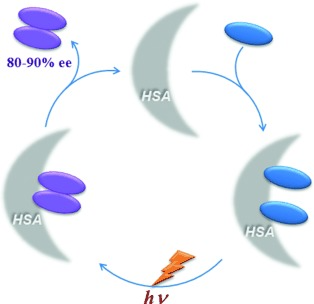 Catalytic Bio-Supramolecular Photochirogenesis: Batch-Operated Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate with Human Serum Albumin.Masaki Nishijima, Hanako Kato, Cheng Yang, Gaku Fukuhara, Tadashi Mori, Yasuyuki Araki, Takehiko Wada, and Yoshihisa Inoue*ChemCatChem, 2013, 5, 1411–1413.
Catalytic Bio-Supramolecular Photochirogenesis: Batch-Operated Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate with Human Serum Albumin.Masaki Nishijima, Hanako Kato, Cheng Yang, Gaku Fukuhara, Tadashi Mori, Yasuyuki Araki, Takehiko Wada, and Yoshihisa Inoue*ChemCatChem, 2013, 5, 1411–1413.Catalytic bio-supramolecular photochirogenesis is achieved for the enantiodifferentiating [4+4] photocyclodimerization of 2-anthracenecarboxylate by repeatedly using human serum albumin (HSA) as a chiral mediator; the original high enantiomeric excess values (up to 80–90%) are preserved.
@article{nishijima2013catalytic, title = {Catalytic Bio-Supramolecular Photochirogenesis: Batch-Operated Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate with Human Serum Albumin.}, author = {Nishijima, Masaki and Kato, Hanako and Yang, Cheng and Fukuhara, Gaku and Mori, Tadashi and Araki, Yasuyuki and Wada, Takehiko and Inoue, Yoshihisa}, journal = {ChemCatChem}, volume = {5}, number = {11}, year = {2013}, doi = {10.1002/cctc.201300160}, url = {https://doi.org/10.1002/cctc.201300160}, dimensions = {true}, tab = {paper}, } -
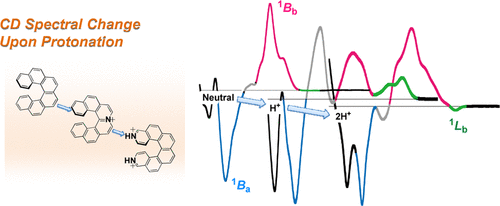 Theoretical and experimental studies of circular dichroism of mono-and diazonia [6] helicenesYoshito Nakai, Tadashi Mori*, Kiyoshi Sato, and Yoshihisa Inoue*J. Phys. Chem. A, 2013, 117, 5082–5092.
Theoretical and experimental studies of circular dichroism of mono-and diazonia [6] helicenesYoshito Nakai, Tadashi Mori*, Kiyoshi Sato, and Yoshihisa Inoue*J. Phys. Chem. A, 2013, 117, 5082–5092.Combined experimental and theoretical studies revealed the characteristic circular dichroism (CD) spectral profiles of mono- and diazonia[6]helicenes, which were distinctly different from those reported for parent [6]helicene and neutral (di)aza-analogues. Aza[6]helicenes and [6]helicene showed bisignate Cotton effects (CEs) at the 1Ba and 1Bb bands, along with a weak CE at the 1Lb band, where the signs of the former bands are responsible for the helical chirality of the helicenes while the sign of the latter is susceptive to the various factors such as electronic and steric effects. Protonation to monoaza[6]helicenes produces azonia[6]helicenes, showing dramatic changes in the CE pattern from the two bisignate to a three positive, two negative CE extremum series of comparable magnitudes, while dual protonation to diaza[6]helicenes forming diazonia[6]helicenes led to only nominal changes (slightly different rotational strength and excitation energy) in the CE pattern. Such rather complicated and contrasting CE behaviors of mono- versus diazoniahelicenes are derived mostly from the electronic effects of (unsymmetrical) protonation because the structures of neutral, mono-, and dicationic species are essentially identical to each other. Compared with those of neutral (di)aza[6]helicenes, the experimental CD spectra of (di)azonia[6]helicenes were less satisfactorily reproduced by the theoretical calculations at the state-of-the-art RI-CC2/TZVPP//DFT-D2-B97-D/TZVP level, most probably due to the inadequate incorporation of the effects of solvation. Nevertheless, the bytheoretical predictions were reasonably accurate and highly valuable in assigning the observed CE and elucidating the origin of the elaborate CD spectral behaviors upon protonation through inspection of the molecular orbital configuration of each transition, encouraging the extended use of the present protocol for analyzing the CD spectral behavior of aza- and other heteroatom-incorporated helicenes upon protonation. The CD spectral behavior upon metal ligation will also be explained through further theoretical and experimental studies.
@article{nakai2013theoretical, title = {Theoretical and experimental studies of circular dichroism of mono-and diazonia [6] helicenes}, author = {Nakai, Yoshito and Mori, Tadashi and Sato, Kiyoshi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. A}, volume = {117}, number = {24}, pages = {5082--5092}, year = {2013}, publisher = {ACS Publications}, doi = {10.1021/jp403426w}, url = {https://doi.org/10.1021/jp403426w}, dimensions = {true}, tab = {paper}, } -
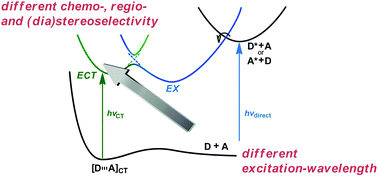 Charge-transfer excitation: unconventional yet practical means for controlling stereoselectivity in asymmetric photoreactionsTadashi Mori* and Yoshihisa InoueChem. Soc. Rev., 2013, 42, 8122–8133.
Charge-transfer excitation: unconventional yet practical means for controlling stereoselectivity in asymmetric photoreactionsTadashi Mori* and Yoshihisa InoueChem. Soc. Rev., 2013, 42, 8122–8133.In chiral donor–acceptor (D–A) systems, irradiation wavelength plays vital roles in determining the photochemical consequences. Selective excitation of a D–A complex at the charge-transfer (C-T) band affords an excited C-T complex (ECT), while the local-band excitation of D or A may lead to the formation of a conventional exciplex (EX) upon subsequent interaction with the D–A partner. These two excited species, generated from the same D–A pair, may be categorized formally as excited complexes or exciplexes, but should be distinguished, provided that they significantly differ in structure and reactivity. Indeed, ECT and EX exhibit distinctly different temperature-dependent photophysical and photochemical behaviours, which are assignable to the differences in relative stability, conformational flexibility and/or solvation properties. Fine-tuning excitation wavelength further enabled us to discriminate stereoisomeric intramolecular C-T complexes through preferential excitation, as C-T complexes are generally composed of an ensemble of various geometries. Besides temperature and solvent polarity, the excitation wavelength was shown to be employed as an unconventional yet practical tool for critically controlling the chemo-, regio- and stereoselectivities in molecular and supramolecular photochemistry.
@article{mori2013charge, title = {Charge-transfer excitation: unconventional yet practical means for controlling stereoselectivity in asymmetric photoreactions}, author = {Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chem. Soc. Rev.}, volume = {42}, number = {20}, pages = {8122--8133}, year = {2013}, publisher = {Royal Society of Chemistry}, doi = {10.1039/C3CS60117J}, url = {https://doi.org/10.1039/C3CS60117J}, dimensions = {true}, tab = {review}, } -
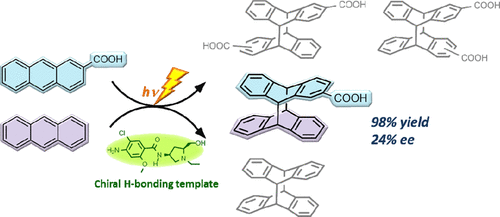 Cross-versus homo-photocyclodimerization of anthracene and 2-anthracenecarboxylic acid mediated by a chiral hydrogen-bonding template. Factors controlling the cross-/homo-selectivity and enantioselectivityYuko Kawanami, Hiroaki Umehara, Jun-ichi Mizoguchi, Masaki Nishijima, Gaku Fukuhara, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2013, 78, 3073–3085.
Cross-versus homo-photocyclodimerization of anthracene and 2-anthracenecarboxylic acid mediated by a chiral hydrogen-bonding template. Factors controlling the cross-/homo-selectivity and enantioselectivityYuko Kawanami, Hiroaki Umehara, Jun-ichi Mizoguchi, Masaki Nishijima, Gaku Fukuhara, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2013, 78, 3073–3085.Competitive cross-/homo-photocyclodimerization of anthracene (AN) and 2-anthracenecarboxylic acid (AC) mediated by a chiral hydrogen-bonding template (TKS) was investigated under various conditions. The cross-photocyclodimerization was favored by a factor of 4–5 at all temperatures and wavelengths examined to afford the AC-AN cross-dimer in 80–84% yield even at AN/AC = 1 and in 98% yield at AN/AC = 10. The enantiomeric excesses (ee’s) obtained were 27–47% for the homo-dimers and 21–24% for the cross-dimer. The absolute configuration of the cross-dimer was determined by comparing the experimental and theoretical circular dichroism spectra and further correlated with the re/si enantiotopic-face selectivity upon AC-TKS complexation in the ground state. Detailed analyses of the complexation behavior and the fluorescence lifetime and cyclodimerization rate of excited re/si complexes revealed that the product’s ee is critically controlled not only by the relative abundance of the re/si complexes in the ground and excited states but also by their relative photocyclodimerization rate. Crucially, the ground-state thermodynamics and the excited-state kinetics are not synergistic but offsetting in enantiotopic-face selectivity, and the latter overwhelms the former to give the homo- and cross-dimers in modest ee’s. Finally, some practical strategies for enhancing the enantioselectivity in chiral template-mediated photochirogenesis have been proposed.
@article{kawanami2013cross, title = {Cross-versus homo-photocyclodimerization of anthracene and 2-anthracenecarboxylic acid mediated by a chiral hydrogen-bonding template. Factors controlling the cross-/homo-selectivity and enantioselectivity}, author = {Kawanami, Yuko and Umehara, Hiroaki and Mizoguchi, Jun-ichi and Nishijima, Masaki and Fukuhara, Gaku and Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {78}, number = {7}, pages = {3073--3085}, year = {2013}, publisher = {ACS Publications}, doi = {10.1021/jo302818w}, url = {https://doi.org/10.1021/jo302818w}, dimensions = {true}, tab = {paper}, } -
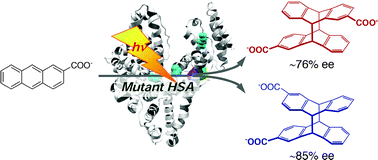 Photochirogenesis with mutant human serum albumins: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylateMasaki Nishijima, Hanako Kato, Gaku Fukuhara, Cheng Yang, Tadashi Mori, Toru Maruyama, Masaki Otagiri*, and Yoshihisa Inoue*Chem. Commun., 2013, 49, 7433–7435.
Photochirogenesis with mutant human serum albumins: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylateMasaki Nishijima, Hanako Kato, Gaku Fukuhara, Cheng Yang, Tadashi Mori, Toru Maruyama, Masaki Otagiri*, and Yoshihisa Inoue*Chem. Commun., 2013, 49, 7433–7435.Mutant human serum albumins accelerated the photocyclodimerization of 2-anthracenecarboxylate to afford chiral cyclodimers in 75–85% enantiomeric excesses, revealing that the mutations to impair non-productive sites 1 and/or 2 enhanced the substrate binding to site 3 without seriously damaging its inherently high photochirogenic ability.
@article{nishijima2013photochirogenesis, title = {Photochirogenesis with mutant human serum albumins: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate}, author = {Nishijima, Masaki and Kato, Hanako and Fukuhara, Gaku and Yang, Cheng and Mori, Tadashi and Maruyama, Toru and Otagiri, Masaki and Inoue, Yoshihisa}, journal = {Chem. Commun.}, volume = {49}, number = {67}, pages = {7433--7435}, year = {2013}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c3cc42656d}, url = {https://doi.org/10.1039/c3cc42656d}, dimensions = {true}, tab = {paper}, } -
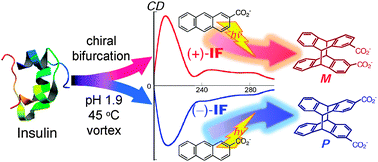 Supramolecular photochirogenesis with functional amyloid superstructuresMasaki Nishijima, Hidekazu Tanaka, Cheng Yang, Gaku Fukuhara, Tadashi Mori, Viktoria Babenko, Wojciech Dzwolak*, and Yoshihisa Inoue*Chem. Commun., 2013, 49, 8916–8918.
Supramolecular photochirogenesis with functional amyloid superstructuresMasaki Nishijima, Hidekazu Tanaka, Cheng Yang, Gaku Fukuhara, Tadashi Mori, Viktoria Babenko, Wojciech Dzwolak*, and Yoshihisa Inoue*Chem. Commun., 2013, 49, 8916–8918.Chiral variants of amyloid fibrils prepared by agitating acidified solutions of bovine insulin at 45 °C not only induced quasi-mirror-imaged circular dichroism spectra upon complexation with 2-anthracenecarboxylate but also gave anti-head-to-head-cyclodimers of the opposite absolute configurations upon photoirradiation.
@article{nishijima2013supramolecular, title = {Supramolecular photochirogenesis with functional amyloid superstructures}, author = {Nishijima, Masaki and Tanaka, Hidekazu and Yang, Cheng and Fukuhara, Gaku and Mori, Tadashi and Babenko, Viktoria and Dzwolak, Wojciech and Inoue, Yoshihisa}, journal = {Chem. Commun.}, volume = {49}, number = {79}, pages = {8916--8918}, year = {2013}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c3cc44235g}, url = {https://doi.org/10.1039/c3cc44235g}, dimensions = {true}, tab = {paper}, } -
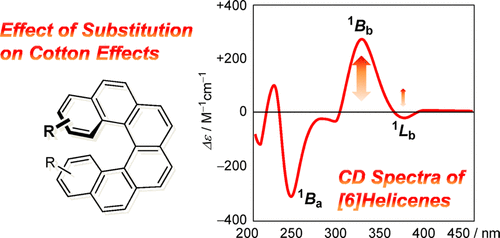 Circular dichroism of (di) methyl-and diaza [6] helicenes. A combined theoretical and experimental studyYoshito Nakai, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. A, 2013, 117, 83–93.
Circular dichroism of (di) methyl-and diaza [6] helicenes. A combined theoretical and experimental studyYoshito Nakai, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. A, 2013, 117, 83–93.Circular dichroism (CD) and relevant chiroptical properties of (di)methyl- and diaza[6]helicenes were investigated by the state-of-the-art approximate coupled cluster and density functional theory calculations, results of which were compared with the corresponding experimental data obtained for newly synthesized enantiopure helicenes. The theoretical calculation at the RI-CC2/TZVPP//DFT-D2-B97-D/TZVP level accurately reproduced the experimental CD spectra in both excitation energy and rotational strength. The electric and magnetic transition dipole moment vectors for the helical sense-responsive 1Bb and the substitution-sensitive 1Lb bands were compared with those for parent carbo[6]helicene, from which the effects of methyl and nitrogen introduced at different positions upon the experimental CD spectra were discussed to separately evaluate the electronic and steric consequences of the substitution to the chiroptical properties. The electronic effects of substitution on CD spectra were further investigated theoretically by employing a series of 3,3-disubstituted [6]helicenes. This first systematic investigation allows us not only to accurately reproduce the experimental CD spectra of known substituted helicenes but also to directly envisage the chiroptical properties of unknown helicenes.
@article{nakai2013circular, title = {Circular dichroism of (di) methyl-and diaza [6] helicenes. A combined theoretical and experimental study}, author = {Nakai, Yoshito and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. A}, volume = {117}, number = {1}, pages = {83--93}, year = {2013}, publisher = {ACS Publications}, doi = {10.1021/jp3104084}, url = {https://doi.org/10.1021/jp3104084}, dimensions = {true}, tab = {paper}, } -
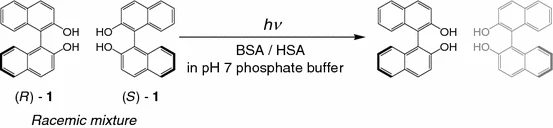 Chiral recognition and supramolecular photoreaction of 1, 1′-binaphthol with bovine and human serum albuminsMasaki Nishijima, Jae-Won Chang, Cheng Yang, Gaku Fukuhara, Tadashi Mori, and Yoshihisa Inoue*Res. Chem. Intermed., 2013, 39, 371–383.
Chiral recognition and supramolecular photoreaction of 1, 1′-binaphthol with bovine and human serum albuminsMasaki Nishijima, Jae-Won Chang, Cheng Yang, Gaku Fukuhara, Tadashi Mori, and Yoshihisa Inoue*Res. Chem. Intermed., 2013, 39, 371–383.In their pioneering study in 1991, Levi-Minzi and Zandomeneghi discovered that photoirradiation of racemic 1,1′-binaphthol (rac-1) in the presence of bovine serum albumin (BSA) in distilled water gave (R)-1 in 99 % enantiomeric excess (ee) after 77 % of the starting material had been consumed. No similar attempt was made with human serum albumin (HSA). In this study of the effects of phosphate buffer solution on the ground-state affinity and excited-state photobehavior of 1 with serum albumin we found that both BSA and HSA preferentially bind the (S) enantiomer of 1 and that photoreaction of rac-1 mediated by BSA and HSA affords (R)-1 in 98 % ee with 99 % conversion and in 46 % ee with 65 % conversion, respectively.
@article{nishijima2013chiral, title = {Chiral recognition and supramolecular photoreaction of 1, 1′-binaphthol with bovine and human serum albumins}, author = {Nishijima, Masaki and Chang, Jae-Won and Yang, Cheng and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Res. Chem. Intermed.}, volume = {39}, number = {1}, pages = {371--383}, year = {2013}, publisher = {Springer}, doi = {10.1007/s11164-012-0655-1}, url = {https://doi.org/10.1007/s11164-012-0655-1}, dimensions = {true}, tab = {paper}, } -
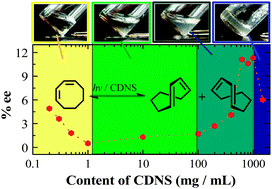 Phase-controlled supramolecular photochirogenesis in cyclodextrin nanospongesWenting Liang, Cheng Yang*, Dayang Zhou, Hitoshi Haneoka, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Franca Castiglione, Andrea Mele*, Fabrizio Caldera, Francesco Trotta*, and Yoshihisa Inoeu*Chem. Commun., 2013, 49, 3510–3512.
Phase-controlled supramolecular photochirogenesis in cyclodextrin nanospongesWenting Liang, Cheng Yang*, Dayang Zhou, Hitoshi Haneoka, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Franca Castiglione, Andrea Mele*, Fabrizio Caldera, Francesco Trotta*, and Yoshihisa Inoeu*Chem. Commun., 2013, 49, 3510–3512.Pyromellitate-bridged cyclodextrin nanosponges (CDNSs) evolved from sol into gel state upon gradual increase of the concentration from 0.2 to 2000 mg mL−1 in water. The enantiodifferentiating geometrical photoisomerizations of (Z)-cyclooctene and (Z,Z)-1,3-cyclooctadiene sensitized by CDNS at various concentrations were critically affected by the phase transition of CDNS to afford the corresponding (E)- and (E,Z)-isomers in the highest enantiomeric excesses in the gel state.
@article{liang2013phase, title = {Phase-controlled supramolecular photochirogenesis in cyclodextrin nanosponges}, author = {Liang, Wenting and Yang, Cheng and Zhou, Dayang and Haneoka, Hitoshi and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Castiglione, Franca and Mele, Andrea and Caldera, Fabrizio and Trotta, Francesco and Inoeu, Yoshihisa}, journal = {Chem. Commun.}, volume = {49}, number = {34}, pages = {3510--3512}, year = {2013}, publisher = {Royal Society of Chemistry}, doi = {10.1039/C3CC40542G}, url = {https://doi.org/10.1039/C3CC40542G}, dimensions = {true}, tab = {paper}, } -
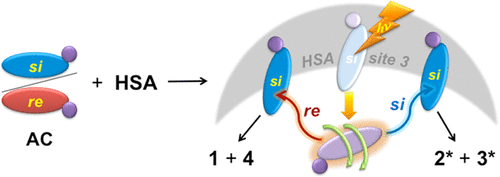 Explaining the highly enantiomeric photocyclodimerization of 2-anthracenecarboxylate bound to human serum albumin using time-resolved anisotropy studiesDenis Fuentealba, Hanako Kato, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cornelia Bohne*J. Am. Chem. Soc., 2013, 135, 203–209.
Explaining the highly enantiomeric photocyclodimerization of 2-anthracenecarboxylate bound to human serum albumin using time-resolved anisotropy studiesDenis Fuentealba, Hanako Kato, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Yoshihisa Inoue*, and Cornelia Bohne*J. Am. Chem. Soc., 2013, 135, 203–209.The mechanism for the high enantiomeric excess (ee) (80–90%) observed in the photocyclodimerization of 2-anthracenecarboxylate (AC) in the chiral binding sites of human serum albumin (HSA) was studied using fluorescence anisotropy. A long rotational correlation time of 36 ns was observed for the excited states of the ACs bound to the HSA site responsible for the high ee, suggesting that the ACs have restricted rotational mobility in this site. The ACs in this site have the same prochiral face protected by the protein, and this protection is responsible for the high ee observed. These insights provide a strategy for the rational design of supramolecular photochirogenic systems.
@article{fuentealba2013explaining, title = {Explaining the highly enantiomeric photocyclodimerization of 2-anthracenecarboxylate bound to human serum albumin using time-resolved anisotropy studies}, author = {Fuentealba, Denis and Kato, Hanako and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa and Bohne, Cornelia}, journal = {J. Am. Chem. Soc.}, volume = {135}, number = {1}, pages = {203--209}, year = {2013}, publisher = {ACS Publications}, doi = {10.1021/ja3081555}, url = {https://doi.org/10.1021/ja3081555}, dimensions = {true}, tab = {paper}, }
2012
-
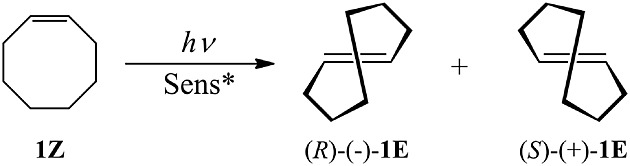 Supramolecular Photochirogenesis with Novel Cyclic Tetrasaccharide: Enantiodifferentiating Photoisomerization of (Z)-Cyclooctene with Cyclic Nigerosylnigerose-Based SensitizersCheng Yang*, Wenting Liang, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Hiroyuki Hiramatsu, Yasufumi Dan-oh, Kazuo Tsujimoto, and Yoshihisa Inoue*Chirality, 2012, 24, 921–927.
Supramolecular Photochirogenesis with Novel Cyclic Tetrasaccharide: Enantiodifferentiating Photoisomerization of (Z)-Cyclooctene with Cyclic Nigerosylnigerose-Based SensitizersCheng Yang*, Wenting Liang, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Hiroyuki Hiramatsu, Yasufumi Dan-oh, Kazuo Tsujimoto, and Yoshihisa Inoue*Chirality, 2012, 24, 921–927.Isophthalic and terephthalic acid monoesters of cyclic nigerosyl-(1→6)-nigerose (CNN), a cyclic tetrasaccharide composed of four d-glucopyranosyl residues connected by alternating α-1,3- and α-1,6-linkages, were synthesized as novel chiral supramolecular sensitizers for enantiodifferentiating photoisomerization of (Z)-cyclooctene (1Z) to planar chiral (E)-isomer (1E). Despite the saucer-shaped shallow cavity of CNN that does not immediately guarantee strong ground-state interactions with 1Z, the sensitizer-appended CNNs afforded optically active 1E in such enantiomeric excesses that are much improved than those obtained with an α-cyclodextrin analog and comparable with those obtained with a β-cyclodextrin analog. Interestingly, the enantiomeric excess values obtained were a critical function of temperature and solvent to show an inversion of the product chirality by changing the environmental variants. Nevertheless, all of the differential activation parameters calculated from the temperature-dependent enantiomeric excesses gave an excellent compensatory enthalpy–entropy relationship, indicating an operation of a single enantiodifferentiating mechanism in the present chiral photosensitization with modified CNNs.
@article{yang2012supramolecular, title = {Supramolecular Photochirogenesis with Novel Cyclic Tetrasaccharide: Enantiodifferentiating Photoisomerization of (Z)-Cyclooctene with Cyclic Nigerosylnigerose-Based Sensitizers}, author = {Yang, Cheng and Liang, Wenting and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Hiramatsu, Hiroyuki and Dan-oh, Yasufumi and Tsujimoto, Kazuo and Inoue, Yoshihisa}, journal = {Chirality}, volume = {24}, number = {11}, pages = {921--927}, year = {2012}, publisher = {Wiley Online Library}, doi = {10.1002/chir.22014}, url = {https://doi.org/10.1002/chir.22014}, dimensions = {true}, tab = {paper}, } -
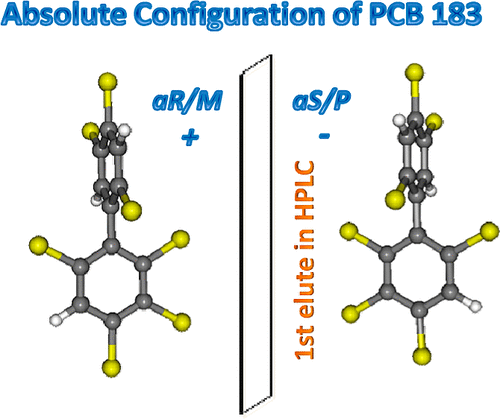 Absolute configuration of atropisomeric polychlorinated biphenyl 183 enantiomerically enriched in human samplesMitsunobu Toda, Chisato Matsumura, Masahiro Tsurukawa, Toshihiro Okuno, Takeshi Nakano*, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2012, 116, 9340–9346.
Absolute configuration of atropisomeric polychlorinated biphenyl 183 enantiomerically enriched in human samplesMitsunobu Toda, Chisato Matsumura, Masahiro Tsurukawa, Toshihiro Okuno, Takeshi Nakano*, Yoshihisa Inoue, and Tadashi Mori*J. Phys. Chem. A, 2012, 116, 9340–9346.Polychlorinated biphenyls (PCBs) are still of serious concern as a potential health hazard due to their persistency and bioacumulation. Of 209 possible PCB congeners, with varying number and position of chlorine atom(s), 19 are chiral. These are mostly highly chlorinated and tend to remain longer against the biological decompositions, suffering biological deracemization in the environment. In this work, we have unequivocally determined the absolute configurations of important chiral PCBs 183 and 171, as well as 132, through the combined theoretical and experimental investigations of the chiroptical properties (circular dichroism and optical rotation), which will be valuable in elucidating the mechanism of biological enantiomer enrichment of PCBs in the environment.
@article{toda2012absolute, title = {Absolute configuration of atropisomeric polychlorinated biphenyl 183 enantiomerically enriched in human samples}, author = {Toda, Mitsunobu and Matsumura, Chisato and Tsurukawa, Masahiro and Okuno, Toshihiro and Nakano, Takeshi and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Phys. Chem. A}, volume = {116}, number = {37}, pages = {9340--9346}, year = {2012}, publisher = {ACS Publications}, doi = {10.1021/jp306363n}, url = {https://doi.org/10.1021/jp306363n}, dimensions = {true}, tab = {paper}, } -
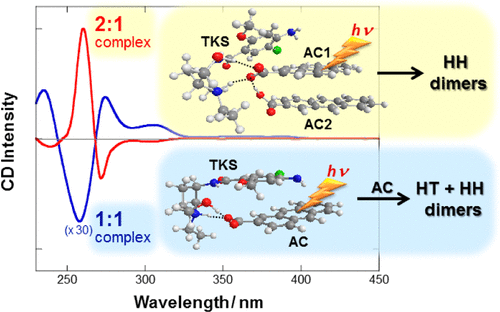 Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid via competitive binary/ternary hydrogen-bonded complexes with 4-benzamidoprolinolYuko Kawanami, Shin-ya Katsumata, Jun-ichi Mizoguchi, Masaki Nishijima, Gaku Fukuhara, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*Org. Lett., 2012, 14, 4962–4965.
Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid via competitive binary/ternary hydrogen-bonded complexes with 4-benzamidoprolinolYuko Kawanami, Shin-ya Katsumata, Jun-ichi Mizoguchi, Masaki Nishijima, Gaku Fukuhara, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*Org. Lett., 2012, 14, 4962–4965.Circular dichroism (CD) spectral examinations at various host/guest ratios revealed that 2-anthracenecarboxylic acid (AC) forms not only 1:1 but also novel 2:1 hydrogen-bonded/π-stacked complexes with a chiral 4-benzamidoprolinol template (TKS159). The 2:1 complexation is a minor process but causes significant CD spectral changes as a consequence of the exciton coupling interaction of two AC chromophores and greatly accelerates the head-to-head photocyclodimerization to significantly affect the stereochemical outcomes.
@article{kawanami2012enantiodifferentiating, title = {Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid via competitive binary/ternary hydrogen-bonded complexes with 4-benzamidoprolinol}, author = {Kawanami, Yuko and Katsumata, Shin-ya and Mizoguchi, Jun-ichi and Nishijima, Masaki and Fukuhara, Gaku and Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {14}, number = {18}, pages = {4962--4965}, year = {2012}, publisher = {ACS Publications}, doi = {10.1021/ol3023402}, url = {https://doi.org/10.1021/ol3023402}, dimensions = {true}, tab = {paper}, } -
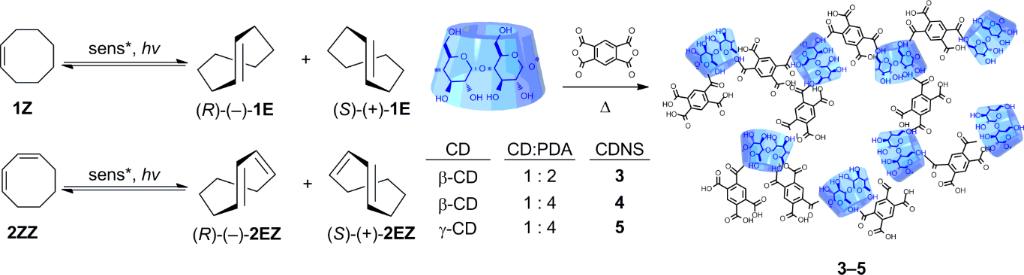 Cyclodextrin nanosponge-sensitized enantiodifferentiating photoisomerization of cyclooctene and 1, 3-cyclooctadieneWenting Liang, Cheng Yang*, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Andrea Mele*, Franca Castiglione, Fabrizio Caldera, Francesco Trotta*, and Yoshihisa Inoue*Beilstein J. Org. Chem., 2012, 8, 1305–1311.
Cyclodextrin nanosponge-sensitized enantiodifferentiating photoisomerization of cyclooctene and 1, 3-cyclooctadieneWenting Liang, Cheng Yang*, Masaki Nishijima, Gaku Fukuhara, Tadashi Mori, Andrea Mele*, Franca Castiglione, Fabrizio Caldera, Francesco Trotta*, and Yoshihisa Inoue*Beilstein J. Org. Chem., 2012, 8, 1305–1311.Enantiodifferentiating geometrical photoisomerizations of (Z)-cyclooctene and (Z,Z)-1,3-cyclooctadiene were performed by using the pyromellitate-linked cyclodextrin network polymer, termed “cyclodextrin nanosponge (CDNS)”, as a supramolecular sensitizing host. The photochirogenic behavior of the nanosponges incorporating β- or γ-cyclodextrin was significantly different from that reported for the conventional sensitizer-appended monomeric cyclodextrins, affording chiral (E)-cyclooctene and (E,Z)-cyclooctadiene in enantiomeric excesses critically dependent on the solution pH and solvent composition employed, revealing the active roles of chiral void spaces of CDNS in the photochirogenic reaction.
@article{liang2012cyclodextrin, title = {Cyclodextrin nanosponge-sensitized enantiodifferentiating photoisomerization of cyclooctene and 1, 3-cyclooctadiene}, author = {Liang, Wenting and Yang, Cheng and Nishijima, Masaki and Fukuhara, Gaku and Mori, Tadashi and Mele, Andrea and Castiglione, Franca and Caldera, Fabrizio and Trotta, Francesco and Inoue, Yoshihisa}, journal = {Beilstein J. Org. Chem.}, volume = {8}, number = {1}, pages = {1305--1311}, year = {2012}, publisher = {Beilstein-Institut}, doi = {10.3762/bjoc.8.149}, url = {https://doi.org/10.3762/bjoc.8.149}, dimensions = {true}, tab = {paper}, } -
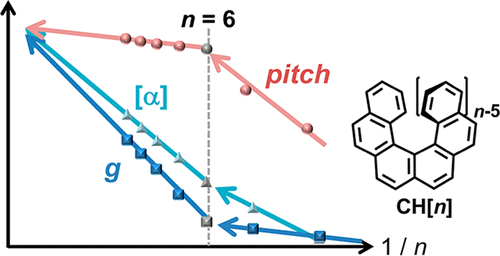 Theoretical and experimental studies on circular dichroism of carbo [n] helicenesYoshito Nakai, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. A, 2012, 116, 7372–7385.
Theoretical and experimental studies on circular dichroism of carbo [n] helicenesYoshito Nakai, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. A, 2012, 116, 7372–7385.The chiroptical properties of a series of carbo[n]helicenes (n = 4–10) were investigated by the state-of-the-art approximate coupled cluster and density functional theory calculations. The theoretical calculation at the RI-CC2/TZVPP//DFT-D2-B97-D/TZVP level nicely reproduced the experimental CD spectra in both excitation energy and rotational strength without any shift or scaling. These calculations afforded the electric and the magnetic transition dipole moment vectors in [n]helicenes, allowing us to discuss the observed rotational strengths as a function of the number of benzene rings. Although the observed CD intensity was not immediately correlated to any of the calculated parameters, the anisotropy (g) factor of the 1Bb band and the specific rotation were found inversely proportional to n and nicely correlated with the helical pitch, but discontinuous at n = 6, where the aromatic rings start to overlap. In contrast, the g factor at the 1Ba band was rather insensitive to n. It was also revealed that the excitation energies of the 1Bb and 1Ba bands are inversely proportional to n over the entire range of n examined. The theoretical predictions also enabled us to rectify the erroneous experimental CD spectra of [5]- and [6]helicenes reported earlier, by using the enantiopure samples resolved by chiral HPLC.
@article{nakai2012theoretical, title = {Theoretical and experimental studies on circular dichroism of carbo [n] helicenes}, author = {Nakai, Yoshito and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. A}, volume = {116}, number = {27}, pages = {7372--7385}, year = {2012}, publisher = {ACS Publications}, doi = {10.1021/jp304576g}, url = {https://doi.org/10.1021/jp304576g}, dimensions = {true}, tab = {paper}, } -
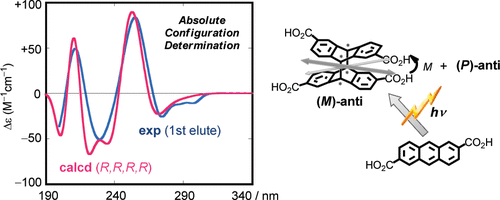 Theoretical and experimental investigations of circular dichroism and absolute configuration determination of chiral anthracene photodimersAyako Wakai, Hiroki Fukasawa, Cheng Yang, Tadashi Mori*, and Yoshihisa Inoue*J. Am. Chem. Soc., 2012, 134, 4990–4997.
Theoretical and experimental investigations of circular dichroism and absolute configuration determination of chiral anthracene photodimersAyako Wakai, Hiroki Fukasawa, Cheng Yang, Tadashi Mori*, and Yoshihisa Inoue*J. Am. Chem. Soc., 2012, 134, 4990–4997.Substituted anthracenes photodimerize to stereoisomeric [4 + 4] cyclodimers, some of which are inherently chiral. Recent supramolecular photochirogenic studies enabled the efficient preparation of specific stereoisomers, the absolute configurations of which should reflect the chiral environment of supramolecular host or scaffold employed but have not been determined, hindering detailed mechanistic elucidation and further host/scaffold design. In this study, we performed the combined experimental and state-of-the-art theoretical analyses of the circular dichroism spectra of chiral cyclodimers of 2-anthracenecarboxylic and 2,6-anthracenedicarboxylic acids to reveal the configurational and molecular orbital origin of the Cotton effects observed, and unambiguously determined the absolute configurations of these chiral cyclodimers. The present results allow us to directly correlate the enantiotopic face-selectivity upon photocyclodimerization with the absolute configuration of the cyclodimer derived therefrom and also to precisely elucidate the chiral arrangement of two cyclodimerizing anthracenes.
@article{wakai2012theoretical, title = {Theoretical and experimental investigations of circular dichroism and absolute configuration determination of chiral anthracene photodimers}, author = {Wakai, Ayako and Fukasawa, Hiroki and Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {134}, number = {10}, pages = {4990--4997}, year = {2012}, publisher = {ACS Publications}, doi = {10.1021/ja300522y}, url = {https://doi.org/10.1021/ja300522y}, dimensions = {true}, tab = {paper}, } -
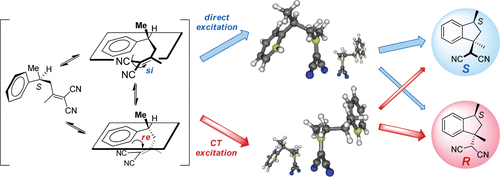 Control of Conformer Population and Product Selectivity and Stereoselectivity in Competitive Photocyclization/Rearrangement of Chiral Donor–Acceptor DyadEmi Nishiuchi, Tadashi Mori*, and Yoshihisa Inoue*Journal of the American Chemical Society, 2012, 134, 8082–8085.
Control of Conformer Population and Product Selectivity and Stereoselectivity in Competitive Photocyclization/Rearrangement of Chiral Donor–Acceptor DyadEmi Nishiuchi, Tadashi Mori*, and Yoshihisa Inoue*Journal of the American Chemical Society, 2012, 134, 8082–8085.The conformer population and the relative excitation efficiency of each conformer, assessed by theoretical and experimental examinations, enabled us to precisely control the chemo- and diastereoselectivities of the competitive photocyclization/rearrangement reaction of a chiral donor–acceptor (D/A) dyad by irradiation wavelength, solvent polarity, and reaction temperature. Manipulating the ground- and excited-state conformer equilibria is essential for critically controlling the intramolecular D/A system, in sharp contrast to the rather divergent excited species involved in relevant intermolecular systems.
@article{nishiuchi2012control, title = {Control of Conformer Population and Product Selectivity and Stereoselectivity in Competitive Photocyclization/Rearrangement of Chiral Donor--Acceptor Dyad}, author = {Nishiuchi, Emi and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Journal of the American Chemical Society}, volume = {134}, number = {19}, pages = {8082--8085}, year = {2012}, publisher = {ACS Publications}, doi = {10.1021/ja302768w}, url = {https://doi.org/10.1021/ja302768w}, dimensions = {true}, tab = {paper}, } -
 Strictly diastereocontrolled photocyclodimerization of 2-anthracenecarboxylates tethered to cyclic tetrasaccharidesGaku Fukuhara*, Tomohiro Nakamura, Yuko Kawanami, Cheng Yang, Tadashi Mori, Hiroyuki Hiramatsu, Yasufumi Dan-Oh, Kazuo Tsujimoto, and Yoshihisa Inoue*Chem. Commun., 2012, 48, 9156–9158.
Strictly diastereocontrolled photocyclodimerization of 2-anthracenecarboxylates tethered to cyclic tetrasaccharidesGaku Fukuhara*, Tomohiro Nakamura, Yuko Kawanami, Cheng Yang, Tadashi Mori, Hiroyuki Hiramatsu, Yasufumi Dan-Oh, Kazuo Tsujimoto, and Yoshihisa Inoue*Chem. Commun., 2012, 48, 9156–9158.Photocyclodimerization of 2-anthracenecarboxylate tethered to a cyclic nigerosylnigerose scaffold gave a single chiral cyclodimer (out of two achiral and two chiral stereoisomers) in 99% optical and 96% chemical yields, achieving the ultimate stereocontrol of the supramolecular photochirogenesis in aqueous solution at 25 °C.
@article{fukuhara2012strictly, title = {Strictly diastereocontrolled photocyclodimerization of 2-anthracenecarboxylates tethered to cyclic tetrasaccharides}, author = {Fukuhara, Gaku and Nakamura, Tomohiro and Kawanami, Yuko and Yang, Cheng and Mori, Tadashi and Hiramatsu, Hiroyuki and Dan-Oh, Yasufumi and Tsujimoto, Kazuo and Inoue, Yoshihisa}, journal = {Chem. Commun.}, volume = {48}, number = {73}, pages = {9156--9158}, year = {2012}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c2cc34880b}, url = {https://doi.org/10.1039/c2cc34880b}, dimensions = {true}, tab = {paper}, } - Accomplishment of Determining Method for Absolute Configuration: Reproduction of CD Spectra by Theoretical CalculationsTadashi Mori and Yoshihisa InoueKagaku, 2012, 67, 72–73.
@article{mori2012accomplishment, title = {Accomplishment of Determining Method for Absolute Configuration: Reproduction of CD Spectra by Theoretical Calculations}, author = {Mori, Tadashi and Inoue, Yoshihisa}, journal = {Kagaku}, volume = {67}, issue = {11}, pages = {72--73}, year = {2012} }
2011
-
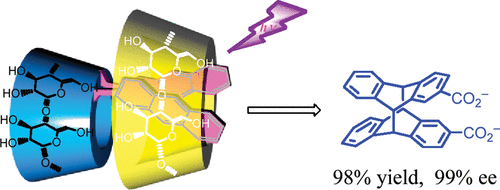 Dual supramolecular photochirogenesis: ultimate stereocontrol of photocyclodimerization by a chiral scaffold and confining hostCheng Yang, Chenfeng Ke, Wenting Liang, Gaku Fukuhara, Tadashi Mori, Yu Liu, and Yoshihisa Inoue*J. Am. Chem. Soc., 2011, 133, 13786–13789.
Dual supramolecular photochirogenesis: ultimate stereocontrol of photocyclodimerization by a chiral scaffold and confining hostCheng Yang, Chenfeng Ke, Wenting Liang, Gaku Fukuhara, Tadashi Mori, Yu Liu, and Yoshihisa Inoue*J. Am. Chem. Soc., 2011, 133, 13786–13789.In contrast to the brilliant success in thermal asymmetric synthesis, precise stereocontrol remains a great challenge in chiral photochemistry because of the lack of effective tools and methodologies for controlling the short-lived, weakly interacting, and highly reactive electronically excited species. In this work, we achieved this goal through the “dual-chiral, dual-supramolecular” photochirogenesis approach, which enabled us to realized dramatic acceleration and perfect stereocontrol in one of the most representative photoreactions. Thus, the [4 + 4] photocyclodimerization of 2-anthracenecarboxylate tethered to an α-cyclodextrin scaffold was accelerated by a γ-cyclodextrin or cucurbit[8]uril host and gave a single enantiomeric cyclodimer (out of four possible chiral and achiral stereoisomers) in up to 98% chemical and 99% optical yield.
@article{yang2011dual, title = {Dual supramolecular photochirogenesis: ultimate stereocontrol of photocyclodimerization by a chiral scaffold and confining host}, author = {Yang, Cheng and Ke, Chenfeng and Liang, Wenting and Fukuhara, Gaku and Mori, Tadashi and Liu, Yu and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {133}, number = {35}, pages = {13786--13789}, year = {2011}, publisher = {ACS Publications}, doi = {10.1021/ja202020x}, url = {https://doi.org/10.1021/ja202020x}, dimensions = {true}, tab = {paper}, } -
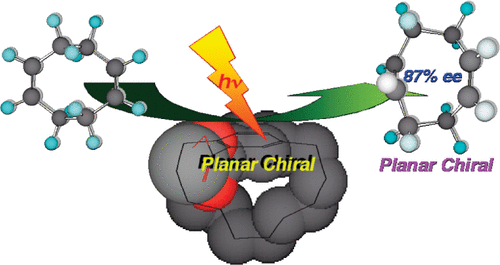 Planar-to-planar chirality transfer in the excited state. Enantiodifferentiating photoisomerization of cyclooctenes sensitized by planar-chiral paracyclophaneRyo Maeda, Takehiko Wada, Tadashi Mori, Shigeyuki Kono, Nobuhiro Kanomata*, and Yoshihisa Inoue*J. Am. Chem. Soc., 2011, 133, 10379–10381.
Planar-to-planar chirality transfer in the excited state. Enantiodifferentiating photoisomerization of cyclooctenes sensitized by planar-chiral paracyclophaneRyo Maeda, Takehiko Wada, Tadashi Mori, Shigeyuki Kono, Nobuhiro Kanomata*, and Yoshihisa Inoue*J. Am. Chem. Soc., 2011, 133, 10379–10381.Photochemical planar-to-planar chirality transfer was effected by using (R)-[10]paracyclophane-12-carboxylates as a planar-chiral sensitizer and (Z)-cyclooctene and (Z,Z)-1,5-cyclooctadiene as prochiral substrates to give a planar-chiral (E)- and (E,Z)-isomer in up to 44% and 87% enantiomeric excess, respectively, the latter of which being the highest ever reported for a sensitized photochirogenic reaction.
@article{maeda2011planar, title = {Planar-to-planar chirality transfer in the excited state. Enantiodifferentiating photoisomerization of cyclooctenes sensitized by planar-chiral paracyclophane}, author = {Maeda, Ryo and Wada, Takehiko and Mori, Tadashi and Kono, Shigeyuki and Kanomata, Nobuhiro and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {133}, number = {27}, pages = {10379--10381}, year = {2011}, publisher = {ACS Publications}, doi = {10.1021/ja203781f}, url = {https://doi.org/10.1021/ja203781f}, dimensions = {true}, tab = {paper}, } -
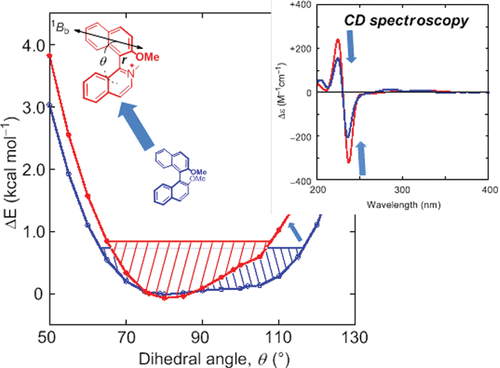 Axial chirality of donor–donor, donor–acceptor, and tethered 1, 1′-binaphthyls: a theoretical revisit with dynamics trajectoriesMasaki Nishizaka, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. A, 2011, 115, 5488–5495.
Axial chirality of donor–donor, donor–acceptor, and tethered 1, 1′-binaphthyls: a theoretical revisit with dynamics trajectoriesMasaki Nishizaka, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. A, 2011, 115, 5488–5495.The circular dichroism (CD) spectra of (R)-2,2′-dimethoxy-1,1′-binaphthyl (DD) and its untethered and tethered donor–acceptor analogues (DA and DA7–DA9) were investigated experimentally and theoretically. The experimental CD spectra of DD and DA resembled each other in several aspects, displaying a positive–positive–negative Cotton effect pattern in the 1Lb–1La region and a strong negative couplet at the 1Bb band, but significantly differed in transition energy and rotatory strength. The couplet amplitude (A) of the main band was 1.6 times larger in DA than in DD, despite the comparable extinction coefficients and seemingly analogous conformations. An additional positive Cotton effect was observed at the CT (CT) band for donor–acceptor binaphthyl DA. Our theoretical prediction of the CD spectra of binaphthyls involves three sequential first principle quantum mechanics (QM) calculations. Thus, the geometry optimizations of a series of conformers with varying dihedral angles were performed by the dispersion-corrected DFT-D method using the B97-D functional and the TZV2P basis set. The potential curve as a function of the dihedral angle (θ) was obtained by using the SCS-MP2/TZVPP single-point energy calculations with and without application of the solvent correction. The CD spectrum of each conformer was independently calculated by the second-order approximate coupled cluster calculation (CC2 method) using the TZVPP basis sets and the resolution of the identity (RI-J) approximation. The (net) theoretical CD spectrum was obtained by averaging over all possible conformers, where the dynamics trajectories based on the relative SCS-MP2 energies were taken into account. By using 17 possible conformers at θ varying from 50 to 130° by 5° intervals, the experimental CD spectra were successfully reproduced in a quantitative manner, enabling us to characterize properly almost all of the important spectral features and chiroptical properties. The two-state model, reported previously, turned out to have led to the right answer with wrong reasons. The couplet sign and amplitude A are critical functions of θ and can be used not only for (qualitatively) determining the absolute configuration but also for quantitatively analyzing the binaphthyl conformations. The angle dependence of A was already argued in the classical coupled oscillator and exciton chirality theories to provide reasonable structure elucidations but only in a qualitative or semiquantitative manner. Our method is able to predict the A value quantitatively as a function of θ. For tethered binaphthyls DA7–DA9, particular care should be exercised in the conformational assessment based on the classical treatment because the amplitude A was shown to be significantly affected by the existence of the tether itself. In the present method, the couplet amplitude A was nicely related to the dihedral angle θ of DA and DD by the state-of-the-art ab initio calculations, enabling us to gain the quantitative information about the conformation of axially chiral binaphthyls. The Cotton effect at the CT band also serves as a complementary clue for elucidating the conformation of donor–acceptor binaphthyls.
@article{nishizaka2011axial, title = {Axial chirality of donor--donor, donor--acceptor, and tethered 1, 1′-binaphthyls: a theoretical revisit with dynamics trajectories}, author = {Nishizaka, Masaki and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. A}, volume = {115}, number = {21}, pages = {5488--5495}, year = {2011}, publisher = {ACS Publications}, doi = {10.1021/jp202776g}, url = {https://doi.org/10.1021/jp202776g}, dimensions = {true}, tab = {paper}, } -
 Enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene included and sensitized by naphthoyl-curdlanGaku Fukuhara*, Mami Imai, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*Org. Lett., 2011, 13, 1856–1859.
Enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene included and sensitized by naphthoyl-curdlanGaku Fukuhara*, Mami Imai, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*Org. Lett., 2011, 13, 1856–1859.6-O-(2-Naphthoyl)curdlan was newly synthesized as a sensitizing polysaccharide host to examine the chiroptical properties, supramolecular complexation, and photochirogenic behavior with (Z,Z)-1,3-cyclooctadiene (1ZZ). The enantiodifferentiating photoisomerization of 1ZZ included and sensitized by this polysaccharide host gave a highly strained chiral (E,Z)-isomer in up to 8.7% enantiomeric excess (ee) in solution and 11.7% ee in the solid state, which are the highest values ever reported for a supramolecular photochirogenesis of 1EZ.
@article{fukuhara2011enantiodifferentiating, title = {Enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene included and sensitized by naphthoyl-curdlan}, author = {Fukuhara, Gaku and Imai, Mami and Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {13}, number = {7}, pages = {1856--1859}, year = {2011}, publisher = {ACS Publications}, doi = {10.1021/ol2003644}, url = {https://doi.org/10.1021/ol2003644}, dimensions = {true}, tab = {paper}, } -
 Supramolecular FRET photocyclodimerization of anthracenecarboxylate with naphthalene-capped γ-cyclodextrinQian Wang, Cheng Yang*, Gaku Fukuhara, Tadashi Mori, Yu Liu, and Yoshihisa Inoue*Beilstein J. Org. Chem., 2011, 7, 290–297.
Supramolecular FRET photocyclodimerization of anthracenecarboxylate with naphthalene-capped γ-cyclodextrinQian Wang, Cheng Yang*, Gaku Fukuhara, Tadashi Mori, Yu Liu, and Yoshihisa Inoue*Beilstein J. Org. Chem., 2011, 7, 290–297.γ-Cyclodextrin (CD) derivatives with a naphthalene moiety anchored to one or two of the glucose units of the CD were synthesized in order to investigate the effects of flexible and rigid capping upon complexation, as well as Förster resonance energy transfer (FRET) and photochirogenic behavior of anthracenecarboxylate (AC) moieties. UV–vis, circular dichroism and fluorescence spectral studies revealed that two AC molecules are simultaneously included in the modified γ-CD cavity to form a right-handed screw and also that the naphthalene cap efficiently transfers the singlet energy to AC included in the CD cavity via the FRET mechanism. Compared to native γ-CD, the modified γ-CDs showed much higher first association constants (K1) but relatively lower second association constants (K2) for AC, leading to two-fold larger overall affinities (K1K2). Photocyclodimerization of AC with these modified γ-CDs produced more head-to-head (HH) dimers in much better enantiomeric excesses (ee) for anti-HH dimer compared to native γ-CD. Interestingly, FRET excitation further enhanced the chemical and optical yields of anti-HH dimer up to 36% and 35% ee, for which the highly efficient FRET sensitization within the CD cavity, minimizing the “contamination” from the achiral “outside” photoreaction, is responsible. FRET sensitization also enabled us to achieve the catalytic photocyclodimerization of AC with a sub-equivalent amount of chiral supramolecular host.
@article{wang2011supramolecular, title = {Supramolecular FRET photocyclodimerization of anthracenecarboxylate with naphthalene-capped $\gamma$-cyclodextrin}, author = {Wang, Qian and Yang, Cheng and Fukuhara, Gaku and Mori, Tadashi and Liu, Yu and Inoue, Yoshihisa}, journal = {Beilstein J. Org. Chem.}, volume = {7}, number = {1}, pages = {290--297}, year = {2011}, publisher = {Beilstein-Institut}, doi = {10.3762/bjoc.7.38}, url = {https://doi.org/10.3762/bjoc.7.38}, dimensions = {true}, tab = {paper}, } -
 Chiral ionic liquid-mediated photochirogenesis. Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acidGaku Fukuhara*, Takahiro Okazaki, Marco Lessi, Masaki Nishijima, Cheng Yang, Tadashi Mori, Andrea Mele, Fabio Bellina, Cinzia Chiappe*, and Yoshihisa Inoue*Org. Biomol. Chem., 2011, 9, 7105–7112.
Chiral ionic liquid-mediated photochirogenesis. Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acidGaku Fukuhara*, Takahiro Okazaki, Marco Lessi, Masaki Nishijima, Cheng Yang, Tadashi Mori, Andrea Mele, Fabio Bellina, Cinzia Chiappe*, and Yoshihisa Inoue*Org. Biomol. Chem., 2011, 9, 7105–7112.Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid (AC-H) and its lithium salt (AC-Li) in chiral ionic liquid (CIL), (R)-1-(2,3-dihydroxypropyl)-3-methylimidazolium acetate [(R)-GLYMI][AcO], gave a mixture of two head-to-tail (HT) and two head-to-head (HH) cyclodimers in HT/HH ratios of 1.3–1.7 (for AC-H) and 2.2–4.3 (for AC-Li) with low enantiomeric excesses (ee) of 0–3% for chiral syn-HT and anti-HH dimers. In contrast, irradiation of AC-H in an aqueous solution, containing cucurbit[8]uril (CB[8]) as a host and [(R)-GLYMI][AcO] or [(R)-GLYMI][Tf2N] as a modifier of CB portals, afforded the HH dimers in 91–99% selectivity, although the anti-HH dimer was totally racemic. Interestingly, irradiation of AC-H in a dichloromethane solution, containing [(R)-GLYMI][AcO] as a chiral template, led to the formation of the HH-dimers in 98% selectivity with chiral anti-HH dimer in −14% ee, presumably by the dual ligation of two ACs to a CIL through electrostatic and hydrogen-bonding interactions.
@article{fukuhara2011chiral, title = {Chiral ionic liquid-mediated photochirogenesis. Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid}, author = {Fukuhara, Gaku and Okazaki, Takahiro and Lessi, Marco and Nishijima, Masaki and Yang, Cheng and Mori, Tadashi and Mele, Andrea and Bellina, Fabio and Chiappe, Cinzia and Inoue, Yoshihisa}, journal = {Org. Biomol. Chem.}, volume = {9}, number = {20}, pages = {7105--7112}, year = {2011}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c1ob05716b}, url = {https://doi.org/10.1039/c1ob05716b}, dimensions = {true}, tab = {paper}, } -
 Competitive photocyclization/rearrangement of 4-aryl-1, 1-dicyanobutenes controlled by intramolecular charge-transfer interaction. Effect of medium polarity, temperature, pressure, excitation wavelength, and confinementTadashi Ito, Emi Nishiuchi, Gaku Fukuhara, Yoshihisa Inoue*, and Tadashi Mori*Photochem. Photobiol. Sci., 2011, 10, 1405–1414.
Competitive photocyclization/rearrangement of 4-aryl-1, 1-dicyanobutenes controlled by intramolecular charge-transfer interaction. Effect of medium polarity, temperature, pressure, excitation wavelength, and confinementTadashi Ito, Emi Nishiuchi, Gaku Fukuhara, Yoshihisa Inoue*, and Tadashi Mori*Photochem. Photobiol. Sci., 2011, 10, 1405–1414.A series of 4-aryl-1,1-dicyanobutenes (1a-1f) with different substituents were synthesized to control the intramolecular donor-acceptor or charge-transfer (C-T) interactions in the ground state. Photoexcitation of these C-T substrates led to competitive cyclization and rearrangement, the ratio being critically controlled by various environmental factors, such as solvent polarity, temperature and static pressure, and also by excitation wavelength and supramolecular confinement (polyethylene voids). In non-polar solvents, the rearrangement was dominant (>10: 1) for all examined substrates, while the cyclization was favoured in polar solvents, in particular at low temperatures. Selective excitation at the C-T band further enhanced the cyclization up to >50: 1 ratios. More importantly, the cyclization/rearrangement ratio was revealed to be a linear function of the C-T transition energy. However, the substrates with a sterically demanding or highly electron-donating substituent failed to give the cyclization product.
@article{ito2011competitive, title = {Competitive photocyclization/rearrangement of 4-aryl-1, 1-dicyanobutenes controlled by intramolecular charge-transfer interaction. Effect of medium polarity, temperature, pressure, excitation wavelength, and confinement}, author = {Ito, Tadashi and Nishiuchi, Emi and Fukuhara, Gaku and Inoue, Yoshihisa and Mori, Tadashi}, journal = {Photochem. Photobiol. Sci.}, volume = {10}, number = {9}, pages = {1405--1414}, year = {2011}, publisher = {Springer}, doi = {10.1039/c1pp05038a}, url = {https://doi.org/10.1039/c1pp05038a}, dimensions = {true}, tab = {paper}, } -
 Supramolecular complexation and photocyclodimerization of methyl 3-methoxy-2-naphthoate with modified γ-cyclodextrinsWenting Liang, Hui-Hui Zhang, Jing-Jing Wang, Yuan Peng, Bin Chen, Chen-Ho Yang, Li-Zhu Wu, Gaku Fukuhara, Tadashi Mori, and Yoshihisa InouePure Appl. Chem., 2011, 83, 769–778.
Supramolecular complexation and photocyclodimerization of methyl 3-methoxy-2-naphthoate with modified γ-cyclodextrinsWenting Liang, Hui-Hui Zhang, Jing-Jing Wang, Yuan Peng, Bin Chen, Chen-Ho Yang, Li-Zhu Wu, Gaku Fukuhara, Tadashi Mori, and Yoshihisa InouePure Appl. Chem., 2011, 83, 769–778.A series of modified γ-cyclodextrins (CDs) were repared as chiral host for catalyzing the enantiodifferentiating photocyclodimerization of methyl 3-methoxy-2-naph-thoate (NA). The complexation behavior of NA with modifiedγ-CDs was studied by UV–vis,fluorescence, and circular dichroism spectroscopies. All of the modified γ-CDs formed stable1:2 host–guest complex with NA, and binding affinities for two-step complexation riticallydepended on the structure of modified γ-CDs. The enantioselectivity of NA photocyclodimerization was also significantly affected by the modification of γ-CD. Thesecondary rim-modification considerably reduced the enantioselectivity, for which the inter-rupted hydrogen-bonding network, leading to a flexible CD skeleton, is most probably responsible.
@article{liang2011supramolecular, title = {Supramolecular complexation and photocyclodimerization of methyl 3-methoxy-2-naphthoate with modified γ-cyclodextrins}, author = {Liang, Wenting and Zhang, Hui-Hui and Wang, Jing-Jing and Peng, Yuan and Chen, Bin and Yang, Cheng an Tung, Chen-Ho and Wu, Li-Zhu and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Pure Appl. Chem.}, volume = {83}, number = {4}, pages = {769--778}, year = {2011}, publisher = {DE Gruyter}, doi = {10.1351/PAC-CON-10-10-04}, url = {https://doi.org/10.1351/PAC-CON-10-10-04}, dimensions = {true}, tab = {paper}, } -
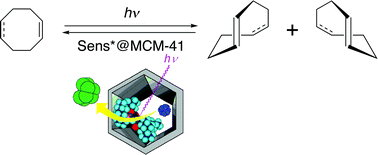 Role of entropy in supramolecular photochirogenesis: Enantiodifferentiating photoisomerization of cyclooctenes in chiral sensitizer-immobilized MCM-41 cavitiesRyo Maeda, Takehiko Wada*, Atsushi Kusaka, Tadashi Mori, Masakazu Iwamoto, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2011, 10, 1390–1392.
Role of entropy in supramolecular photochirogenesis: Enantiodifferentiating photoisomerization of cyclooctenes in chiral sensitizer-immobilized MCM-41 cavitiesRyo Maeda, Takehiko Wada*, Atsushi Kusaka, Tadashi Mori, Masakazu Iwamoto, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2011, 10, 1390–1392.To examine the effects of confinement, or low-entropy environments, we employed the mesoporous silicate MCM-41 modified with optically active benzenetetracarboxylates as chiral reaction media to effect the enantiodifferentiating photoisomerization of (Z)-cyclooctene and (Z,Z)-1,5-cyclooctadiene. The ee of the (E)-isomer produced and its temperature dependence behavior in MCM-41 were completely different from those observed in homogeneous solutions. Immobilizing the sensitizer in MCM-41 reduced the contribution of the entropy factor.
@article{maeda2011role, title = {Role of entropy in supramolecular photochirogenesis: Enantiodifferentiating photoisomerization of cyclooctenes in chiral sensitizer-immobilized MCM-41 cavities}, author = {Maeda, Ryo and Wada, Takehiko and Kusaka, Atsushi and Mori, Tadashi and Iwamoto, Masakazu and Inoue, Yoshihisa}, journal = {Photochem. Photobiol. Sci.}, volume = {10}, number = {9}, pages = {1390--1392}, year = {2011}, publisher = {Springer}, doi = {10.1039/c1pp05087g}, url = {https://doi.org/10.1039/c1pp05087g}, dimensions = {true}, tab = {paper}, } -
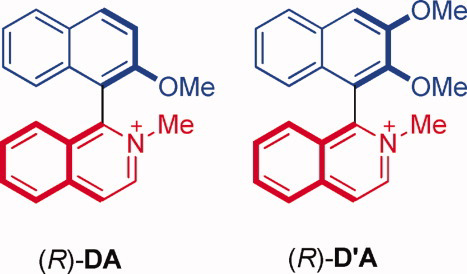 Experimental and theoretical investigations of circular dichroism of donor–acceptor 1, 1′-binaphthyls: Influence of substitution on the coupling amplitude and cotton effect of the charge-transfer bandYoshito Nakai, Masaki Nishizaka, Cheng Yang, Gaku Fukuhara, Tadashi Mori*, and Yoshihisa InoueChirality, 2011, 23, E22–E27.
Experimental and theoretical investigations of circular dichroism of donor–acceptor 1, 1′-binaphthyls: Influence of substitution on the coupling amplitude and cotton effect of the charge-transfer bandYoshito Nakai, Masaki Nishizaka, Cheng Yang, Gaku Fukuhara, Tadashi Mori*, and Yoshihisa InoueChirality, 2011, 23, E22–E27.The electronic circular dichroism (CD) spectra of donor–acceptor binaphthyls were investigated experimentally and theoretically. The enantiomerically pure forms of 1-(2-methoxy-1-naphthyl)- and 1-(2,3-dimethoxy-1-naphthyl)-2-methylisoquinolinium tetrafluoroborates (DA and D′A) were prepared, and their UV–vis and CD spectra were compared. The donor–acceptor interaction was apparent from the absorption at longer wavelengths, whereas its strength was not very different from each other. In addition, very similar structures were obtained for the two aromatic planes in DA and D′A when the geometry was optimized by the density functional theory. The additional methoxy group in the latter spices scarcely disturbed the UV–vis spectrum but significantly affected the CD spectrum. Thus, the observed CD spectra were considerably different from each other, especially in the 1Bb band couplet, where the amplitude was reduced to almost one-fourth in D′A. The theoretical investigations led to the following conclusions: (1) The potential curve associated with the central CC dihedral angle of 1,1′-binaphthyl is fairly flat at the bottom for both DA and D′A and freely rotating at an ambient temperature. The potential curve of D′A is, however, significantly different from that of DA, in which the curve is much steeper and biased to the s-cis side. As the observed CD spectrum is an ensemble of conformers of various dihedral angles, such difference in potential certainly affects the overall spectrum; (2) The additional methoxy group introduced at the 3-position effectively altered the CD spectral pattern, which was theoretically supported by the calculation at the RI-CC2/TZVPP level; (3) Consequently, the classical coupled oscillator theory, in which the angle between the transition dipole moments of two aromatic planes is solely considered, is not applicable to the quantitative evaluation of the chiroptical properties of 1,1′-binaphthyls; rather, the quantum chemical approach is preferred, permitting a direct comparison with the experiment.
@article{nakai2011experimental, title = {Experimental and theoretical investigations of circular dichroism of donor--acceptor 1, 1′-binaphthyls: Influence of substitution on the coupling amplitude and cotton effect of the charge-transfer band}, author = {Nakai, Yoshito and Nishizaka, Masaki and Yang, Cheng and Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chirality}, volume = {23}, number = {1E}, pages = {E22--E27}, year = {2011}, publisher = {Wiley Online Library}, doi = {10.1002/chir.20947}, url = {https://doi.org/10.1002/chir.20947}, dimensions = {true}, tab = {paper}, } -
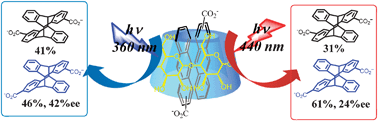 Wavelength-controlled supramolecular photocyclodimerization of anthracenecarboxylate mediated by γ-cyclodextrinsQian Wang, Cheng Yang*, Chengfeng Ke, Gaku Fukuhara, Tadashi Mori, Yu Liu*, and Yoshihisa Inoue*Chem. Commun., 2011, 47, 6849–6851.
Wavelength-controlled supramolecular photocyclodimerization of anthracenecarboxylate mediated by γ-cyclodextrinsQian Wang, Cheng Yang*, Chengfeng Ke, Gaku Fukuhara, Tadashi Mori, Yu Liu*, and Yoshihisa Inoue*Chem. Commun., 2011, 47, 6849–6851.Stereochemical outcomes were critically tuned by excitation wavelength in the supramolecular photocyclodimerization of 2-anthracenecarboxylic acid mediated by native and diamino-modified γ-cyclodextrins.
@article{wang2011wavelength, title = {Wavelength-controlled supramolecular photocyclodimerization of anthracenecarboxylate mediated by γ-cyclodextrins}, author = {Wang, Qian and Yang, Cheng and Ke, Chengfeng and Fukuhara, Gaku and Mori, Tadashi and Liu, Yu and Inoue, Yoshihisa}, journal = {Chem. Commun.}, volume = {47}, number = {24}, pages = {6849--6851}, year = {2011}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c1cc11771h}, url = {https://doi.org/10.1039/c1cc11771h}, dimensions = {true}, tab = {paper}, } -
 Introduction to the themed issue in honour of the contribution of Japanese scientists to photochemistryCornelia Bohne and Tadashi MoriPhotochem. Photobiol. Sci., 2011, 10, 1379–1379.
Introduction to the themed issue in honour of the contribution of Japanese scientists to photochemistryCornelia Bohne and Tadashi MoriPhotochem. Photobiol. Sci., 2011, 10, 1379–1379.A graphical abstract is available for this content
@article{bohne2011introduction, title = {Introduction to the themed issue in honour of the contribution of Japanese scientists to photochemistry}, author = {Bohne, Cornelia and Mori, Tadashi}, journal = {Photochem. Photobiol. Sci.}, volume = {10}, number = {9}, pages = {1379--1379}, year = {2011}, publisher = {Springer}, doi = {10.1039/c1pp90023d}, url = {https://doi.org/10.1039/c1pp90023d}, dimensions = {true}, tab = {paper}, }
2010
-
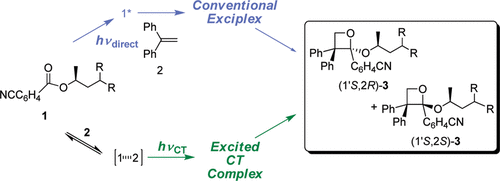 Solvent and Temperature Effects on Diastereodifferentiating Paternò- Büchi Reaction of Chiral Alkyl Cyanobenzoates with Diphenylethene upon Direct versus Charge-Transfer ExcitationKazuyuki Matsumura, Tadashi Mori*, and Yoshihisa InoueJ. Org. Chem., 2010, 75, 5461–5469.
Solvent and Temperature Effects on Diastereodifferentiating Paternò- Büchi Reaction of Chiral Alkyl Cyanobenzoates with Diphenylethene upon Direct versus Charge-Transfer ExcitationKazuyuki Matsumura, Tadashi Mori*, and Yoshihisa InoueJ. Org. Chem., 2010, 75, 5461–5469.In the Paternó−Büchi reaction of chiral p-cyanobenzoates (1) with 1,1-diphenylethene (2), we revealed that the excited charge-transfer (CT) complex formed upon selective excitation at the CT band is distinctly different in structure and reactivity from the conventional exciplex generated through the direct excitation of acceptor 1 which subsequently associates with donor 2. Thus, the favored diastereoface upon photocycloaddition, as well as the temperature- and solvent-dependent behavior of the product’s diastereoselectivity, were highly contrasting, often opposite, to each other upon direct versus CT excitation. From the activation parameters obtained by the Eyring analyses of the diastereoselectivity, we are able to infer that the conventional exciplex is relatively flexible and susceptible to the environmental variants, whereas the CT complex is better π−π stacked and more rigid in the ground state and also in the excited state, leading to the significantly smaller differential activation enthalpies and entropies. More interestingly, the signs of the differential activation parameters determined for direct and CT excitation are consistently opposite to each other and the isokinetic temperatures calculated therefrom differ significantly, unambiguously revealing the distinctly different nature in structure and reactivity of these two excited-state complex species. Thus, the combined use of irradiation wavelength, temperature, and solvent provides us with a convenient, powerful tool not only for elucidating the mechanistic details of photoreaction but also for critically controlling the stereochemical outcomes of photochirogenic reaction.
@article{matsumura2010solvent, title = {Solvent and Temperature Effects on Diastereodifferentiating Paternò- Büchi Reaction of Chiral Alkyl Cyanobenzoates with Diphenylethene upon Direct versus Charge-Transfer Excitation}, author = {Matsumura, Kazuyuki and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {75}, number = {16}, pages = {5461--5469}, year = {2010}, publisher = {ACS Publications}, doi = {10.1021/jo101332x}, url = {https://doi.org/10.1021/jo101332x}, dimensions = {true}, tab = {paper}, } -
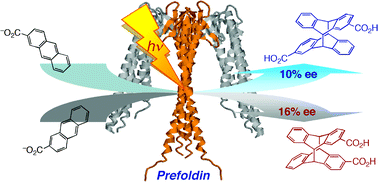 Bio-supramolecular photochirogenesis with molecular chaperone: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by prefoldinKazuki Bando, Tamotsu Zako*, Masafumi Sakono, Mizuo Maeda, Takehiko Wada, Masaki Nishijima, Gaku Fukuhara, Cheng Yang, Tadashi Mori, Tamara CS Pace, Cornelia Bohne*, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2010, 9, 655–660.
Bio-supramolecular photochirogenesis with molecular chaperone: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by prefoldinKazuki Bando, Tamotsu Zako*, Masafumi Sakono, Mizuo Maeda, Takehiko Wada, Masaki Nishijima, Gaku Fukuhara, Cheng Yang, Tadashi Mori, Tamara CS Pace, Cornelia Bohne*, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2010, 9, 655–660.Photocyclodimerization of 2-anthracenecarboxylate mediated by molecular chaperone protein was performed for the first time to afford chiral syn-head-to-tail and anti-head-to-head dimers (2 and 3) in 10% and 16% enantiomeric excess, respectively, with enhanced yields of sterically and electrostatically less-favored head-to-head dimers (3 and 4).
@article{bando2010bio, title = {Bio-supramolecular photochirogenesis with molecular chaperone: enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by prefoldin}, author = {Bando, Kazuki and Zako, Tamotsu and Sakono, Masafumi and Maeda, Mizuo and Wada, Takehiko and Nishijima, Masaki and Fukuhara, Gaku and Yang, Cheng and Mori, Tadashi and Pace, Tamara CS and Bohne, Cornelia and Inoue, Yoshihisa}, journal = {Photochem. Photobiol. Sci.}, volume = {9}, number = {5}, pages = {655--660}, year = {2010}, publisher = {Springer}, doi = {10.1039/B9PP00186G}, url = {https://doi.org/10.1039/B9PP00186G}, dimensions = {true}, tab = {paper}, } -
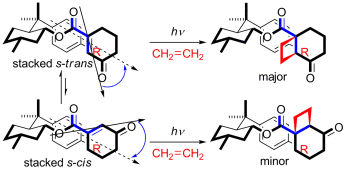 Diastereodifferentiating the [2+ 2] Photocycloaddition of Ethylene to Arylmenthyl Cyclohexenonecarboxylates: Stacking-Driven Enhancement of the Product Diastereoselectivity That Is Correlated with the Reactant EllipticityKen Tsutsumi, Yuuki Yanagisawa, Akinori Furutani, Tsumoru Morimoto, Kiyomi Kakiuchi*, Takehiko Wada, Tadashi Mori, and Yoshihisa Inoue*Chem. Eur. J., 2010, 16, 7448–7455.
Diastereodifferentiating the [2+ 2] Photocycloaddition of Ethylene to Arylmenthyl Cyclohexenonecarboxylates: Stacking-Driven Enhancement of the Product Diastereoselectivity That Is Correlated with the Reactant EllipticityKen Tsutsumi, Yuuki Yanagisawa, Akinori Furutani, Tsumoru Morimoto, Kiyomi Kakiuchi*, Takehiko Wada, Tadashi Mori, and Yoshihisa Inoue*Chem. Eur. J., 2010, 16, 7448–7455.Upon diastereodifferentiating the [2+2] photocycloaddition of ethylene to a series of p-substituted (−)-8-phenylmenthyl cyclohexenonecarboxylates, the diastereoselectivity was critically controlled by the nature of the substituent introduced to the chiral auxiliary, and the p-nitro-substituted substrate afforded the cycloadducts in 90% diastereomeric excess (de) and with 97% isolated yield. Detailed experimental and theoretical conformation analyses revealed that the stacking interaction of the aromatic auxiliary with the cyclohexenone moiety plays the decisive role in determining the substrate conformation and is, therefore, responsible for the dramatic enhancement of the de. Of particular interest, the product de was directly related to the ellipticity of the substrate, enabling us to “predict” the de prior to photoirradiation.
@article{tsutsumi2010diastereodifferentiating, title = {Diastereodifferentiating the [2+ 2] Photocycloaddition of Ethylene to Arylmenthyl Cyclohexenonecarboxylates: Stacking-Driven Enhancement of the Product Diastereoselectivity That Is Correlated with the Reactant Ellipticity}, author = {Tsutsumi, Ken and Yanagisawa, Yuuki and Furutani, Akinori and Morimoto, Tsumoru and Kakiuchi, Kiyomi and Wada, Takehiko and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chem. Eur. J.}, volume = {16}, number = {25}, pages = {7448--7455}, year = {2010}, publisher = {Wiley Online Library}, doi = {10.1002/chem.201000429}, url = {https://doi.org/10.1002/chem.201000429}, dimensions = {true}, tab = {paper}, } -
 Critical stereocontrol by inter-amino distance of supramolecular photocyclodimerization of 2-anthracenecarboxylate mediated by 6-(ω-aminoalkylamino)-γ-cyclodextrinsChenfeng Ke, Cheng Yang, Wenting Liang, Tadashi Mori, Yu Liu*, and Yoshihisa Inoue*New J. Chem., 2010, 34, 1323–1329.
Critical stereocontrol by inter-amino distance of supramolecular photocyclodimerization of 2-anthracenecarboxylate mediated by 6-(ω-aminoalkylamino)-γ-cyclodextrinsChenfeng Ke, Cheng Yang, Wenting Liang, Tadashi Mori, Yu Liu*, and Yoshihisa Inoue*New J. Chem., 2010, 34, 1323–1329.A series of 6-(ω-aminoalkylamino)-6-deoxy-γ-cyclodextrins (CDs) 5–8 with varying inter-amino distances were synthesized to control the stereoselectivity of [4+4] photocyclodimerization of 2-anthracenecarboxylate (AC). Complexation behavior of these CD hosts with AC was studied in aqueous solutions by UV-vis and circular dichroism spectral titration. Supramolecular photocyclodimerization of AC mediated by the CDs was performed in water as well as in water–methanol mixture to reveal that aminopropylamino-CD 6 leads to the formation of head-to-head photodimers 3 and 4 in highest yields, while aminobutylamino-CD 7 affords chiral syn-head-to-tail and anti-head-to-head photodimers 2 and 3 in highest enantioselectivities. These results indicate that the inter-amino distance critically control the product’s stereo- and enantioselectivities through the electrostatic interactions of two anionic AC guests with the dicationic sidechain attached to the γ-CD rim and can be used as a convenient, yet effective, tool for manipulating the stereochemical outcomes of supramolecular photochirogenesis.
@article{ke2010critical, title = {Critical stereocontrol by inter-amino distance of supramolecular photocyclodimerization of 2-anthracenecarboxylate mediated by 6-(ω-aminoalkylamino)-γ-cyclodextrins}, author = {Ke, Chenfeng and Yang, Cheng and Liang, Wenting and Mori, Tadashi and Liu, Yu and Inoue, Yoshihisa}, journal = {New J. Chem.}, volume = {34}, number = {7}, pages = {1323--1329}, year = {2010}, publisher = {Royal Society of Chemistry}, doi = {10.1039/c0nj00131g}, url = {https://doi.org/10.1039/c0nj00131g}, dimensions = {true}, tab = {paper}, } -
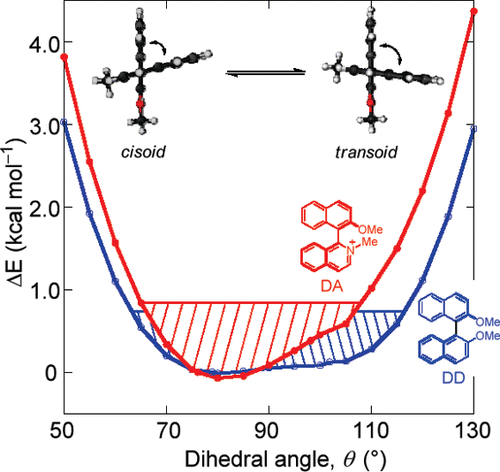 Experimental and Theoretical Studies on the Chiroptical Properties of Donor- Acceptor Binaphthyls. Effects of Dynamic Conformer Population on Circular DichroismMasaki Nishizaka, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. Lett., 2010, 1, 1809–1812.
Experimental and Theoretical Studies on the Chiroptical Properties of Donor- Acceptor Binaphthyls. Effects of Dynamic Conformer Population on Circular DichroismMasaki Nishizaka, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. Lett., 2010, 1, 1809–1812.The axial chirality of biaryls has attracted much attention as effective chiral auxiliaries and ligands in asymmetric synthesis and chirality sensing. In this study, the chiroptical properties of donor−acceptor binaphthyl DA and symmetrical DD were investigated. The amplitude of the main-band couplet was enhanced in the circular dichroism (CD) spectra of DA, due to the effective cation−π interactions altering the potential energy profile against the rotational angle about the binaphthyl’s C1−C1′ bond. A shallow unsymmetrical potential curve was obtained for DA, which is in contrast to the almost symmetrical double-well potential for DD. The theoretical calculations of the CD spectra at the RI-CC2 level using the two-state model composed of the s-cis and s-trans conformations successfully reproduce the experimental CD. Dynamic conformer distribution over a wide range of dihedral angle in the chiral binaphthyls was shown to be critical in discussing the performance of such axially chiral molecules.
@article{nishizaka2010experimental, title = {Experimental and Theoretical Studies on the Chiroptical Properties of Donor- Acceptor Binaphthyls. Effects of Dynamic Conformer Population on Circular Dichroism}, author = {Nishizaka, Masaki and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. Lett.}, volume = {1}, number = {12}, pages = {1809--1812}, year = {2010}, publisher = {ACS Publications}, doi = {10.1021/jz100574e}, url = {https://doi.org/10.1021/jz100574e}, dimensions = {true}, tab = {paper}, } -
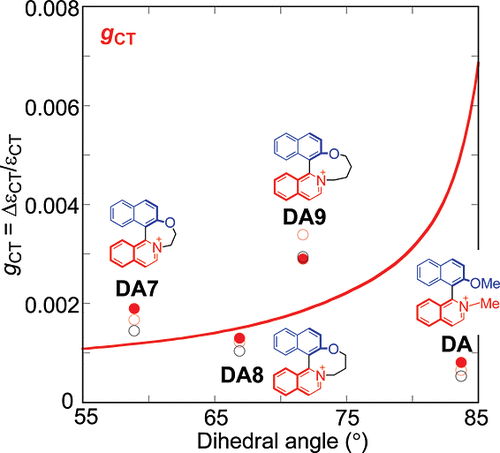 Conformation Elucidation of Tethered Donor- Acceptor Binaphthyls from the Anisotropy Factor of a Charge-Transfer BandMasaki Nishizaka, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. Lett., 2010, 1, 2402–2405.
Conformation Elucidation of Tethered Donor- Acceptor Binaphthyls from the Anisotropy Factor of a Charge-Transfer BandMasaki Nishizaka, Tadashi Mori*, and Yoshihisa Inoue*J. Phys. Chem. Lett., 2010, 1, 2402–2405.Biaryls, in particular 1,1’-binaphthyls, are widely employed as effective chiral auxiliaries and ligands in asymmetric synthesis and chirality sensing, where the amplitude and splitting energy of the main-band couplet are often used as conventional tools for elucidating the conformational details of biaryls in conjunction with relatively simple theoretical models. In this study, the chiroptical properties of a series of tethered donor−acceptor binaphthyls DAn were investigated. The theoretical calculations at the RI-CC2 level successfully reproduced the experimental spectra of all the examined DAn and revealed that the above parameters are quantitatively less informative in the conformational investigations. Alternatively, the anisotropy factor at the charge transfer (CT) band was found to be a more sensitive and robust parameter that can be exploited as a tool for analyzing the solution-phase conformation of donor−acceptor binaphthyls. A substantial contribution from the linker atoms on the anisotropy factor of the CT band was also highlighted.
@article{nishizaka2010conformation, title = {Conformation Elucidation of Tethered Donor- Acceptor Binaphthyls from the Anisotropy Factor of a Charge-Transfer Band}, author = {Nishizaka, Masaki and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. Lett.}, volume = {1}, number = {15}, pages = {2402--2405}, year = {2010}, publisher = {ACS Publications}, doi = {10.1021/jz100901n}, url = {https://doi.org/10.1021/jz100901n}, dimensions = {true}, tab = {paper}, } -
 Circular dichroism of donor–acceptor cyclophanes:(4Rp; 12Rp)-and (4Sp, 12Rp)-12, 15-dimethoxy [2.2] paracyclophane-4, 7-dicarboxylic acid derivativesTakahiro Furo, Tadashi Mori*, and Yoshihisa Inoue*Chirality, 2010, 22, E17–E21.
Circular dichroism of donor–acceptor cyclophanes:(4Rp; 12Rp)-and (4Sp, 12Rp)-12, 15-dimethoxy [2.2] paracyclophane-4, 7-dicarboxylic acid derivativesTakahiro Furo, Tadashi Mori*, and Yoshihisa Inoue*Chirality, 2010, 22, E17–E21.The electronic circular dichroism (CD) spectra were recorded for three diastereomeric eclipsed–staggered pairs of charge-transfer cyclophanes with different substituents, i.e., (4Rp;12Rp)- and (4Sp;12Rp)-12,15-dimethoxy[2.2]paracyclophane-4,7-dicarboxylic acid derivatives (1a-c and 2a-c, where a, b, and c denote methyl ester, carboxylic acid, and carboxylate, respectively). The effects of altering the donor–acceptor interaction between the π systems on the chiroptical properties were experimentally investigated. The anisotropy (g) factors of eclipsed species 2 were not significantly affected by the sort of substituent, while staggered 1 behaved in a significantly different way in CD spectra depending on the charge, affording contrasting CD profiles for neutral a/b versus anionic c. This study provides not only the novel insights into the planar chirality of substituted [2.2]paracyclophanes but also a basis for the potential application of such dramatic CD spectral difference between the acid–base pair to the chiroptical pH-sensors. Chirality 2010. © 2010 Wiley-Liss, Inc.
@article{furo2010circular, title = {Circular dichroism of donor--acceptor cyclophanes:(4Rp; 12Rp)-and (4Sp, 12Rp)-12, 15-dimethoxy [2.2] paracyclophane-4, 7-dicarboxylic acid derivatives}, author = {Furo, Takahiro and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chirality}, volume = {22}, number = {1E}, pages = {E17--E21}, year = {2010}, publisher = {Wiley Online Library}, doi = {10.1002/chir.20835}, url = {https://doi.org/10.1002/chir.20835}, dimensions = {true}, tab = {paper}, } -
 Dual chiral, dual supramolecular diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate tethered to amylose scaffoldGaku Fukuhara*, Tomohiro Nakamura, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*Org. Lett., 2010, 12, 3510–3513.
Dual chiral, dual supramolecular diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate tethered to amylose scaffoldGaku Fukuhara*, Tomohiro Nakamura, Cheng Yang, Tadashi Mori, and Yoshihisa Inoue*Org. Lett., 2010, 12, 3510–3513.Newly synthesized 6-O-(2-anthroyl)amylose (AC-Am; 51% substitution) was photolyzed in (aqueous) DMSO solutions to give HH dimers as major products (after saponification), with modest enantiomeric excesses (ee) of 12−15% and 1−2% for syn-HT and anti-HH dimers, respectively. Addition of γ-cyclodextrin switched the product selectivity to HT and enhanced the ee of syn-HT up to 37%, while the chiral sense of anti-HH was inverted by changing the irradiation temperature, demonstrating usefulness of the dual-supramolecular approach to photochirogenesis.
@article{fukuhara2010dual, title = {Dual chiral, dual supramolecular diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate tethered to amylose scaffold}, author = {Fukuhara, Gaku and Nakamura, Tomohiro and Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {12}, number = {15}, pages = {3510--3513}, year = {2010}, publisher = {ACS Publications}, doi = {10.1021/ol101362s}, url = {https://doi.org/10.1021/ol101362s}, dimensions = {true}, tab = {paper}, } -
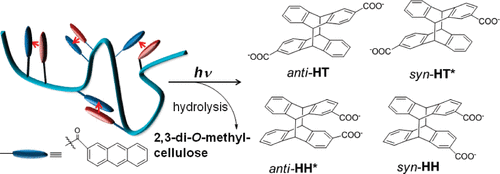 Diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate tethered to cellulose scaffoldGaku Fukuhara*, Tomohiro Nakamura, Cheng Yang, Tadashi Mori, and Yoshihisa InoueJ. Org. Chem., 2010, 75, 4307–4310.
Diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate tethered to cellulose scaffoldGaku Fukuhara*, Tomohiro Nakamura, Cheng Yang, Tadashi Mori, and Yoshihisa InoueJ. Org. Chem., 2010, 75, 4307–4310.A series of anthracenecarboxylate(AC)-appended 2,3-di-O-methylcelluloses (AC-Cells) of varying degrees of substitution (DS) were synthesized to examine their photochirogenic behavior under a variety of conditions. The product distribution and enantiomeric excess of the cyclodimers obtained upon photoirradiation and the subsequent saponification were critical functions of the DS and conversion, for which a conformational change of the flexible polymer backbone is likely to be responsible.
@article{fukuhara2010diastereodifferentiating, title = {Diastereodifferentiating photocyclodimerization of 2-anthracenecarboxylate tethered to cellulose scaffold}, author = {Fukuhara, Gaku and Nakamura, Tomohiro and Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {75}, number = {12}, pages = {4307--4310}, year = {2010}, publisher = {ACS Publications}, doi = {10.1021/jo100596n}, url = {https://doi.org/10.1021/jo100596n}, dimensions = {true}, tab = {paper}, }
2009
-
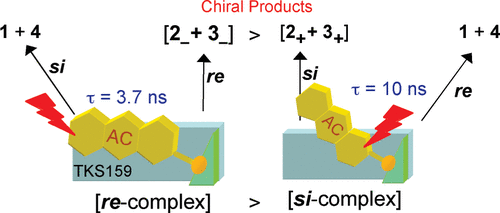 Supramolecular complexation and enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid with 4-aminoprolinol derivatives as chiral hydrogen-bonding templatesYuko Kawanami, Tamara CS Pace, Jun-ichi Mizoguchi, Toshiharu Yanagi, Masaki Nishijima, Tadashi Mori, Takehiko Wada, Cornelia Bohne*, and Yoshihisa Inoue*J. Org. Chem., 2009, 74, 7908–7921.
Supramolecular complexation and enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid with 4-aminoprolinol derivatives as chiral hydrogen-bonding templatesYuko Kawanami, Tamara CS Pace, Jun-ichi Mizoguchi, Toshiharu Yanagi, Masaki Nishijima, Tadashi Mori, Takehiko Wada, Cornelia Bohne*, and Yoshihisa Inoue*J. Org. Chem., 2009, 74, 7908–7921.The photochirogenesis of 2-anthracenecarboxylic acid (AC) complexed to a hydrogen-bonding template (TKS159) was investigated to obtain mechanistic information on how chirogenesis is achieved for the dimerization of AC. Complexation of AC to TKS159 leads to the shielding of one of the two surfaces of the prochiral AC molecule. The two diastereomeric AC−TKS complexes, i.e., re-AC−TKS and si-AC−TKS, were characterized by changes in the UV−vis, fluorescence, and circular dichroism spectra and excited-state lifetimes. The ee is not simply determined by the diastereomeric ratio of the re- and si-AC−TKS complexes but also depends on the relative lifetimes of the diastereomeric complexes. The relative population of the re and si complexes was calculated from the enantiomeric excess (ee) for the products, taking into account the relative lifetimes of the two complexes. These studies established a protocol that can be used to reveal the mechanism for photochirogenesis by investigating the ground state and the excited state behavior of supramolecular systems.
@article{kawanami2009supramolecular, title = {Supramolecular complexation and enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid with 4-aminoprolinol derivatives as chiral hydrogen-bonding templates}, author = {Kawanami, Yuko and Pace, Tamara CS and Mizoguchi, Jun-ichi and Yanagi, Toshiharu and Nishijima, Masaki and Mori, Tadashi and Wada, Takehiko and Bohne, Cornelia and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {74}, number = {20}, pages = {7908--7921}, year = {2009}, publisher = {ACS Publications}, doi = {10.1021/jo901792t}, url = {https://doi.org/10.1021/jo901792t}, dimensions = {true}, tab = {paper}, } -
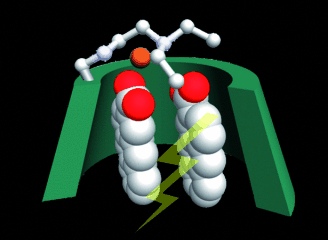 Catalytic enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid mediated by a non-sensitizing chiral metallosupramolecular hostChenfeng Ke, Cheng Yang, Tadashi Mori, Takehiko Wada, Yu Liu*, and Yoshihisa Inoue*Angew. Chem. Int. Ed., 2009, 121, 6803–6805.
Catalytic enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid mediated by a non-sensitizing chiral metallosupramolecular hostChenfeng Ke, Cheng Yang, Tadashi Mori, Takehiko Wada, Yu Liu*, and Yoshihisa Inoue*Angew. Chem. Int. Ed., 2009, 121, 6803–6805.Combined use of diamino-γ-cyclodextrin (CD) and Cu(ClO4)2 resulted in the first catalytic supramolecular photochirogenesis in the photocyclodimerization of 2-anthracenecarboxylic acid. The anti-head-to-head cyclodimer formed in 64–70% enantiomeric excess and about 50% yield; these values are the highest ever reported for CD-mediated photochirogenesis.
@article{ke2009catalytic, title = {Catalytic enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid mediated by a non-sensitizing chiral metallosupramolecular host}, author = {Ke, Chenfeng and Yang, Cheng and Mori, Tadashi and Wada, Takehiko and Liu, Yu and Inoue, Yoshihisa}, journal = {Angew. Chem. Int. Ed.}, volume = {121}, number = {36}, pages = {6803--6805}, year = {2009}, publisher = {WILEY-VCH Verlag Weinheim}, doi = {10.1002/anie.200902911}, url = {https://doi.org/10.1002/anie.200902911}, dimensions = {true}, tab = {paper}, } -
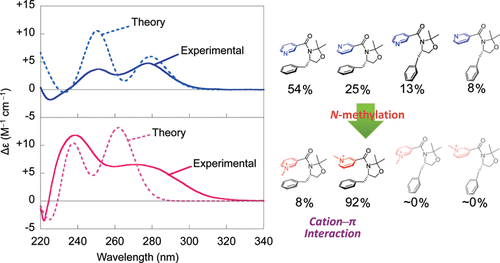 Combined experimental and quantum chemical investigation of chiroptical properties of nicotinamide derivatives with and without intramolecular cation- \pi interactionsAkinori Shimizu, Tadashi Mori*, Yoshihisa Inoue*, and Shinji YamadaJ. Phys. Chem. A, 2009, 113, 8754–8764.
Combined experimental and quantum chemical investigation of chiroptical properties of nicotinamide derivatives with and without intramolecular cation- \pi interactionsAkinori Shimizu, Tadashi Mori*, Yoshihisa Inoue*, and Shinji YamadaJ. Phys. Chem. A, 2009, 113, 8754–8764.The circular dichroism (CD) spectra of neutral and cationic nicotinamide derivatives were experimentally examined in solution and in the solid state to show dramatic differences in the two phases and appreciable dependence on temperature and solvent. The CD spectrum of neutral nicotinamide 1 in solution was reproduced theoretically by averaging the theoretical spectra calculated for all of the extended and folded conformers (s-trans-G+, s-cis-G+, s-trans-T, and s-cis-T) weighted by their population. The preference for the folded, over the extended, conformers in less polar solvent was indicated by the calculation and confirmed experimentally by the analysis of specific rotations. Theoretical CD spectrum calculated for the conformer found in the X-ray structural analysis (s-cis-T) well reproduced the experimental CD spectrum measured in the solid state. Introducing cation−π interactions by N-methylation of 1 to give 1-Me+ led to dramatic changes in CD spectrum. Nevertheless, the experimental CD spectrum of 1-Me+ was well reproduced by averaging the theoretical spectra calculated for a pair of most stable conformers (s-cis-G+ and s-trans-G+) of 1-Me+. The CD spectrum calculated for the s-trans-G+ conformer, which was found in the X-ray crystallographic analysis, did not agree with the experimental one. The theoretical spectra were better reproduced in general by the more sophisticated RI-CC2 method, but the conventional TD-DFT method also gave acceptable results. This allowed us to successfully calculate the larger derivative 2-Me+, for which the RI-CC2 method was not practically applicable. These results show that the structure/conformation may vary with the conditions employed (e.g., by altering the solvent or phase) and thus the experimental analysis under the identical condition is essential for a serious structural study. The present study on a series of nicotinamide derivatives 1, 1-Me+, and 2-Me+ with and without cation−π interactions demonstrates that the combination of experimental and theoretical chiroptical methods is capable of providing reliable structural/conformational information in solution phase, which is complementary to the X-ray crystallographic structure in the solid state.
@article{shimizu2009combined, title = {Combined experimental and quantum chemical investigation of chiroptical properties of nicotinamide derivatives with and without intramolecular cation- $\pi$ interactions}, author = {Shimizu, Akinori and Mori, Tadashi and Inoue, Yoshihisa and Yamada, Shinji}, journal = {J. Phys. Chem. A}, volume = {113}, number = {30}, pages = {8754--8764}, year = {2009}, publisher = {ACS Publications}, doi = {10.1021/jp904243w}, url = {https://doi.org/10.1021/jp904243w}, dimensions = {true}, tab = {paper}, } -
 Supramolecular Complexation of N-Alkyl-and N, N′-Dialkylpiperazines with Cucurbit [6] uril in Aqueous Solution and in the Solid StateMikhail V Rekharsky, Hatsuo Yamamura, Tadashi Mori, Akihiro Sato, Motoo Shiro, Sergey V Lindeman, Rajendra Rathore, Kouhei Shiba, Young Ho Ko, Narayanan Selvapalam, Kimoon Kim*, and Yoshihisa Inoue*Chem. Eur. J., 2009, 15, 1957–1965.
Supramolecular Complexation of N-Alkyl-and N, N′-Dialkylpiperazines with Cucurbit [6] uril in Aqueous Solution and in the Solid StateMikhail V Rekharsky, Hatsuo Yamamura, Tadashi Mori, Akihiro Sato, Motoo Shiro, Sergey V Lindeman, Rajendra Rathore, Kouhei Shiba, Young Ho Ko, Narayanan Selvapalam, Kimoon Kim*, and Yoshihisa Inoue*Chem. Eur. J., 2009, 15, 1957–1965.Complex stoichiometry/composition and degree of oligomerization (oligomeric supramolecular complex formation) of cucurbit[6]uril (CB[6]) with N-alkyl- and N,N′-dialkylpiperazine were investigated in aqueous solutions by means of isothermal titration calorimetry (ITC), ESI-MS, NMR and light scattering measurements. It was found that the complex stability and the degree of oligomerization increase with elongating the alkyl chain attached to the piperazine core. X-ray crystallographic studies revealed a clear correlation between the structure of CB[6]–alkylpiperazine crystals obtained from aqueous solutions and the molecular weight/properties of host–guest oligomers existed in the solution as supramolecular “seeds” of crystal formation.
@article{rekharsky2009supramolecular, title = {Supramolecular Complexation of N-Alkyl-and N, N′-Dialkylpiperazines with Cucurbit [6] uril in Aqueous Solution and in the Solid State}, author = {Rekharsky, Mikhail V and Yamamura, Hatsuo and Mori, Tadashi and Sato, Akihiro and Shiro, Motoo and Lindeman, Sergey V and Rathore, Rajendra and Shiba, Kouhei and Ko, Young Ho and Selvapalam, Narayanan and Kim, Kimoon and Inoue, Yoshihisa}, journal = {Chem. Eur. J.}, volume = {15}, number = {8}, pages = {1957--1965}, year = {2009}, publisher = {Wiley Online Library}, doi = {10.1002/chem.200800398}, url = {https://doi.org/10.1002/chem.200800398}, dimensions = {true}, tab = {paper}, } -
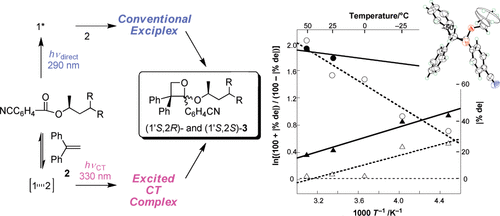 Wavelength Control of Diastereodifferentiating Paternò- Büchi Reaction of Chiral Cyanobenzoates with Diphenylethene through Direct versus Charge-Transfer ExcitationKazuyuki Matsumura, Tadashi Mori, and Yoshihisa InoueJ. Am. Chem. Soc., 2009, 131, 17076–17077.
Wavelength Control of Diastereodifferentiating Paternò- Büchi Reaction of Chiral Cyanobenzoates with Diphenylethene through Direct versus Charge-Transfer ExcitationKazuyuki Matsumura, Tadashi Mori, and Yoshihisa InoueJ. Am. Chem. Soc., 2009, 131, 17076–17077.In the diastereodifferentiating Paternó−Büchi reaction, the excited CT complex was distinctly different in structure and reactivity from the conventional exciplex, and the inherent diastereofacial selectivity and its temperature dependence were opposite to each other in these two excitation modes. Thus, the combined use of wavelength and temperature not only reveals the mechanistic details but also provides a new convenient, powerful tool for critically controlling the stereochemical outcomes of asymmetric photoreactions.
@article{matsumura2009wavelength, title = {Wavelength Control of Diastereodifferentiating Paternò- Büchi Reaction of Chiral Cyanobenzoates with Diphenylethene through Direct versus Charge-Transfer Excitation}, author = {Matsumura, Kazuyuki and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {131}, number = {47}, pages = {17076--17077}, year = {2009}, publisher = {ACS Publications}, doi = {10.1021/ja907156j}, url = {https://doi.org/10.1021/ja907156j}, dimensions = {true}, tab = {paper}, } -
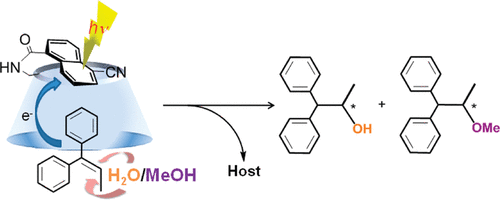 Competitive enantiodifferentiating anti-Markovnikov photoaddition of water and methanol to 1, 1-diphenylpropene using a sensitizing cyclodextrin hostGaku Fukuhara, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2009, 74, 6714–6727.
Competitive enantiodifferentiating anti-Markovnikov photoaddition of water and methanol to 1, 1-diphenylpropene using a sensitizing cyclodextrin hostGaku Fukuhara, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2009, 74, 6714–6727.UV−vis, circular dichroism (CD), fluorescence, and NMR spectral studies on the self-inclusion behavior of a newly synthesized sensitizing host, 6-(5-cyanonaphthyl-1-carboamido)-6-deoxy-β-cyclodextrin (1), showed that the appended naphthalene moiety of 1 perches laterally on the cyclodextrin rim in aqueous methanol but is shallowly included and somewhat tilted in its own cavity in water. UV−vis and CD spectral examinations of the complexation of guest substrate 1,1-diphenylpropene (DPP) with host 1 revealed the formation of a stoichiometric 1:1 complex of DPP with 1. The naphthyl fluorescence of 1 was efficiently quenched by the addition of DPP in aqueous solutions of low methanol contents (≤25%) but was less efficiently quenched in more hydrophobic solvents (≥50% methanol), where the fluorophore is not included in the cavity and allows the external attack of DPP to form an exciplex in the bulk solution. Upon irradiation in aqueous solutions of different methanol contents, competitive photoaddition of water and methanol to DPP occurred to give chiral water adduct 3 and methanol adduct 4, favoring the latter product by a factor of 2.5 due to the higher nucleophilicity of methanol. The enantiomeric excess (ee) values of the photoadducts were generally low in highly methanolic solutions, but was greatly improved by increasing the water content to reach 18% ee for 3 and 13% ee for 4 in 10% methanol solution at −10 °C. Interestingly, the ee of methanol adduct 4 was consistently lower than that of water adduct 3 particularly in water-rich solvents, revealing that the product’s ee is not a simple thermodynamic function of the enantioface-selectivity upon complexation of DPP by chiral host 1 but also kinetically controlled by the subsequent photoinduced enantioface-differentiating nucleophilic attack of water and methanol to radical cationic DPP generated photochemically. Compatible with this mechanism, the compensation plot of the differential activation enthalpy versus entropy, which were obtained from the van’t Hoff analysis of the temperature-dependent ee’s obtained in aqueous solutions of varying methanol contents, gave an excellent straight line for water adduct 3 but an unprecedented bent plot for methanol adduct 4, indicating a switching of the mechanism in between 35 and 50% methanol solution. By using high pressure, low temperature, and/or added salt, the ee of water adduct 3 was further enhanced to 24−26%.
@article{fukuhara2009competitive, title = {Competitive enantiodifferentiating anti-Markovnikov photoaddition of water and methanol to 1, 1-diphenylpropene using a sensitizing cyclodextrin host}, author = {Fukuhara, Gaku and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {74}, number = {17}, pages = {6714--6727}, year = {2009}, publisher = {ACS Publications}, doi = {10.1021/jo9012628}, url = {https://doi.org/10.1021/jo9012628}, dimensions = {true}, tab = {paper}, }
2008
-
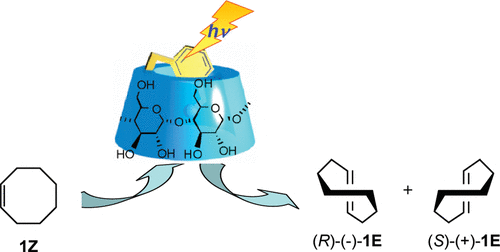 Enantiodifferentiating photoisomerization of cyclooctene included and sensitized by aroyl-β-cyclodextrins: a critical enantioselectivity control by substituentsRunhua Lu*, Cheng Yang, Yujuan Cao, Linhui Tong, Wei Jiao, Takehiko Wada, Zhizhong Wang, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2008, 73, 7695–7701.
Enantiodifferentiating photoisomerization of cyclooctene included and sensitized by aroyl-β-cyclodextrins: a critical enantioselectivity control by substituentsRunhua Lu*, Cheng Yang, Yujuan Cao, Linhui Tong, Wei Jiao, Takehiko Wada, Zhizhong Wang, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2008, 73, 7695–7701.A series of 6-O-benzoyl-β-cyclodextrins (CDs) with methyl, methoxy, methoxycarbonyl, and bromo substituent(s) at the ortho-, meta-, and/or para-position(s) were synthesized as chiral sensitizing hosts for the use in supramolecular enantiodifferentiating photoisomerization of (Z)-cyclooctene (1Z) to its chiral (E)-isomer (1E). The complex stability constants (KS) of the modified β-CDs with 1Z in aqueous methanol solutions were highly sensitive to the substituent(s) introduced to the benzoate moiety, and the log KS values decreased linearly with slightly different slopes with increasing methanol content. The enantiomeric excess (ee) of 1E obtained upon photosensitization with these hosts was a critical function of the size and position of the substituent, varying from almost zero to 46% ee. This indicates that even an apparently small structural difference between methyl and methoxy can critically affect the enantiodifferentiating photoisomerization of a guest included in the chiral cavity, probably through manipulation of both the orientation of ground-state 1Z and the subsequent rotational relaxation of excited 1Z inside the chiral cavity.
@article{lu2008enantiodifferentiating, title = {Enantiodifferentiating photoisomerization of cyclooctene included and sensitized by aroyl-β-cyclodextrins: a critical enantioselectivity control by substituents}, author = {Lu, Runhua and Yang, Cheng and Cao, Yujuan and Tong, Linhui and Jiao, Wei and Wada, Takehiko and Wang, Zhizhong and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {73}, number = {19}, pages = {7695--7701}, year = {2008}, publisher = {ACS Publications}, doi = {10.1021/jo801439n}, url = {https://doi.org/10.1021/jo801439n}, dimensions = {true}, tab = {paper}, } -
 Supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by capped γ-cyclodextrins: critical control of enantioselectivity by cap rigidityCheng Yang, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2008, 73, 5786–5794.
Supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by capped γ-cyclodextrins: critical control of enantioselectivity by cap rigidityCheng Yang, Tadashi Mori, and Yoshihisa Inoue*J. Org. Chem., 2008, 73, 5786–5794.A series of γ-cyclodextrins (CDs) modified with capping and noncapping aromatic group(s) were synthesized to mediate the enantiodifferentiating [4 + 4] photocyclodimerization of 2-anthracenecarboxylic acid (AC). The complexation behavior of these γ-CDs with AC was studied by circular dichroism, UV−vis, and NMR spectroscopy to reveal the formation of stable 1:2 host−guest complexes in all cases. The capped γ-CD with a biphenyl group bridging the A and D glucose units was shown to confine the included AC molecules most strictly among the capped and noncapped γ-CDs examined. Photocyclodimerization of AC mediated by capped γ-CDs considerably improved the yield and enantiomeric excess (ee) of the head-to-head photodimer 3. The ee and the absolute configuration of syn-head-to-tail photodimer 2 critically depended on the rigidity of capping. Thus, the flexibly capped and rim-substituted γ-CDs afforded 2 in moderate ee’s of around 40%, whereas γ-CD with a rigid biphenyl cap gave the antipodal 2 in −58% ee. Interestingly, the ee of 2 mediated by flexibly capped γ-CDs was highly sensitive to the temperature variation as a consequence of large differential entropy changes in the enantiodifferentiation process. In contrast, the entropy effect does not appear to play a significant role in the photocyclodimerization of AC with rigidly capped γ-CDs. The differential enthalpy and entropy changes obtained for the enantiodifferentiating photocyclodimerization mediated by native and most of the modified γ-CDs gave an excellent enthalpy−entropy compensation plot with an exception of the biphenyl-capped γ-CD, indicating the operation of significantly different enantiodifferentiation mechanism within the rigidly capped cyclodextrin cavity.
@article{yang2008supramolecular, title = {Supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate mediated by capped γ-cyclodextrins: critical control of enantioselectivity by cap rigidity}, author = {Yang, Cheng and Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {73}, number = {15}, pages = {5786--5794}, year = {2008}, publisher = {ACS Publications}, doi = {10.1021/jo800533y}, url = {https://doi.org/10.1021/jo800533y}, dimensions = {true}, tab = {paper}, } -
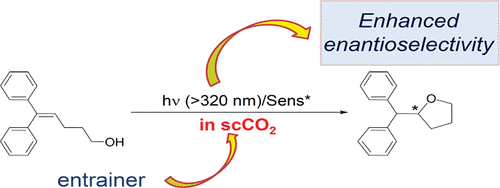 Entrainer effect on photochirogenesis in near-and supercritical carbon dioxide: Dramatic enhancement of enantioselectivityYasuhiro Nishiyama, Takehiko Wada, Sadayuki Asaoka, Tadashi Mori, Taylor A McCarty, Nadine D Kraut, Frank V Bright, and Yoshihisa Inoue*J. Am. Chem. Soc., 2008, 130, 7526–7527.
Entrainer effect on photochirogenesis in near-and supercritical carbon dioxide: Dramatic enhancement of enantioselectivityYasuhiro Nishiyama, Takehiko Wada, Sadayuki Asaoka, Tadashi Mori, Taylor A McCarty, Nadine D Kraut, Frank V Bright, and Yoshihisa Inoue*J. Am. Chem. Soc., 2008, 130, 7526–7527.Diethyl ether added as an entrainer (cosolvent) to near- and supercritical CO2 significantly enhanced the enantioselectivity of photocyclization of 5,5-diphenyl-4-penten-1-ol sensitized by saccharide naphthalenedicarboxylate to give a cyclization product in enantiomeric excesses much larger than those obtained in conventional organic solvents, revealing the unique features of nc- and sc-CO2 as well as the critical role of entrainer clustering to the intervening diastereomeric exciplex pair.
@article{nishiyama2008entrainer, title = {Entrainer effect on photochirogenesis in near-and supercritical carbon dioxide: Dramatic enhancement of enantioselectivity}, author = {Nishiyama, Yasuhiro and Wada, Takehiko and Asaoka, Sadayuki and Mori, Tadashi and McCarty, Taylor A and Kraut, Nadine D and Bright, Frank V and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {130}, number = {24}, pages = {7526--7527}, year = {2008}, publisher = {ACS Publications}, doi = {10.1021/ja801254z}, url = {https://doi.org/10.1021/ja801254z}, dimensions = {true}, tab = {paper}, } -
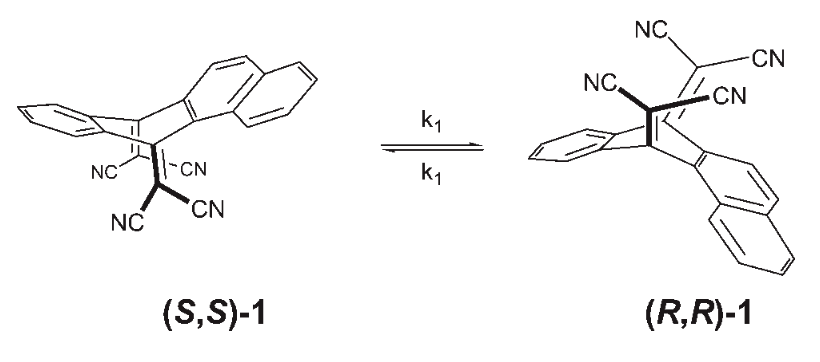 Chiroptical properties and racemization behavior of highly distorted donor-acceptor tetracyanoanthraquinodimethane with interconvertible planar chiralityHideaki Saito, Tadashi Mori*, Yumi Origane, Takehiko Wada, and Yoshihisa InoueChirality: The Pharmacological, Biological, and Chemical Consequences of Molecular Asymmetry, 2008, 20, 278–281.
Chiroptical properties and racemization behavior of highly distorted donor-acceptor tetracyanoanthraquinodimethane with interconvertible planar chiralityHideaki Saito, Tadashi Mori*, Yumi Origane, Takehiko Wada, and Yoshihisa InoueChirality: The Pharmacological, Biological, and Chemical Consequences of Molecular Asymmetry, 2008, 20, 278–281.Absolute configuration of optically active 7,12-bis(dicyanomethylene)-7,12-dihydrobenz[a]anthracene (1) with interconvertible planar chirality was determined by a comparison of experimental and theoretical circular dichroism (CD) spectra. Upon standing at ambient temperatures, 1 was found to spontaneously racemize, the rates of which were assessed through the separation profiles of chiral HPLC at 5–20°C and also from the CD spectral decay profiles at the same temperatures. Mechanism of the racemization was discussed.
@article{saito2008chiroptical, title = {Chiroptical properties and racemization behavior of highly distorted donor-acceptor tetracyanoanthraquinodimethane with interconvertible planar chirality}, author = {Saito, Hideaki and Mori, Tadashi and Origane, Yumi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chirality: The Pharmacological, Biological, and Chemical Consequences of Molecular Asymmetry}, volume = {20}, number = {3-4}, pages = {278--281}, year = {2008}, publisher = {Wiley Online Library}, doi = {10.1002/chir.20427}, url = {https://doi.org/10.1002/chir.20427}, dimensions = {true}, tab = {paper}, } -
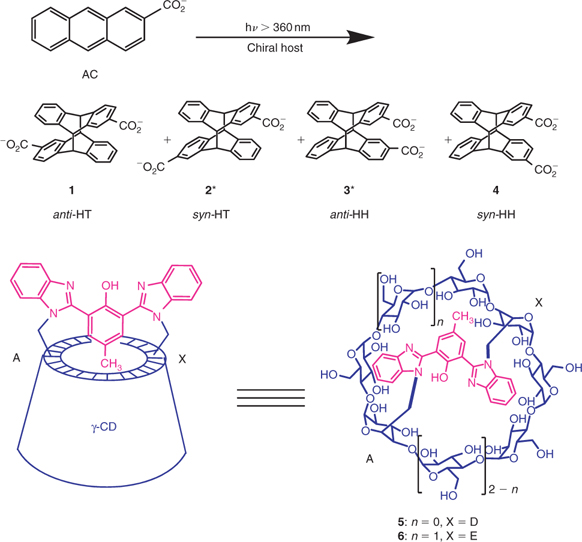 pH-Controlled supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate with capped γ-cyclodextrinsCheng Yang, Chenfeng Ke, Fujita Kahee, De-Qi Yuan*, Tadashi Mori, and Yoshihisa Inoue*Aust. J. Chem., 2008, 61, 565–568.
pH-Controlled supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate with capped γ-cyclodextrinsCheng Yang, Chenfeng Ke, Fujita Kahee, De-Qi Yuan*, Tadashi Mori, and Yoshihisa Inoue*Aust. J. Chem., 2008, 61, 565–568.γ-Cyclodextrins capped with p-cresolbisbenzimidazole at the primary rim were synthesized and used as chiral hosts for mediating the enantiodifferentiating [4 + 4] photocyclodimerization of 2-anthracenecarboxylate. The use of 6A,6E-capped γ-cyclodextrin led to the formation of the anti-head-to-head photodimer in –5% enantiomeric excesses (ee) at pH 11 but in 28% ee at pH 6 with accompanying switching of product chirality, for which a pH-responsive conformational change of the capping moiety is likely to be responsible.
@article{yang2008ph, title = {pH-Controlled supramolecular enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate with capped $\gamma$-cyclodextrins}, author = {Yang, Cheng and Ke, Chenfeng and Kahee, Fujita and Yuan, De-Qi and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Aust. J. Chem.}, volume = {61}, number = {8}, pages = {565--568}, year = {2008}, publisher = {CSIRO Publishing}, doi = {10.1071/CH08143}, url = {https://doi.org/10.1071/CH08143}, dimensions = {true}, tab = {paper}, } -
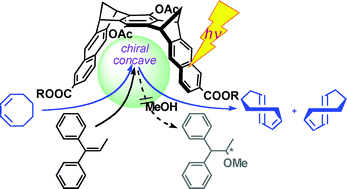 Supramolecular complexation and photochirogenesis with inherently chiral molecular clip: enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene and polar photoaddition to 1, 1-diphenylpropeneGaku Fukuhara, Frank-Gerrit Klärner*, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2008, 7, 1493–1500.
Supramolecular complexation and photochirogenesis with inherently chiral molecular clip: enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene and polar photoaddition to 1, 1-diphenylpropeneGaku Fukuhara, Frank-Gerrit Klärner*, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2008, 7, 1493–1500.Inherently chiral molecular clip (MC) 2 binds (Z,Z)-1,3-cyclooctadiene (COD) and 1,1-diphenylpropene (DPP) in 4:1:5 THF-MeOH-H2O solution (at 25 °C) with association constants of 8800 and 27000 M−1, respectively. The thermodynamic parameters obtained from the van’t Hoff analysis (ΔH° = −96.4 kJ mol−1, ΔS° = −239 J mol−1K−1) reveal that the binding of DPP by MC is strongly driven by the enthalpic gain from hydrophobic and π–π stacking interactions, which is however largely cancelled out by the entropic loss arising from the tight molecular association. Supramolecular photosensitization by MC 2 facilitates the Z–Eisomerization of COD to chiral (E,Z)-isomer in a good E/Z ratio of 0.19 and a low ee of 0.7%, but does not appear to work with DPP probably due to the less-efficient electron transfer in the acceptor–donor–acceptor complex of DPP with MC 2.
@article{fukuhara2008supramolecular, title = {Supramolecular complexation and photochirogenesis with inherently chiral molecular clip: enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene and polar photoaddition to 1, 1-diphenylpropene}, author = {Fukuhara, Gaku and Klärner, Frank-Gerrit and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Photochem. Photobiol. Sci.}, volume = {7}, number = {12}, pages = {1493--1500}, year = {2008}, publisher = {Springer}, doi = {10.1039/b812186a}, url = {https://doi.org/10.1039/b812186a}, dimensions = {true}, tab = {paper}, } -
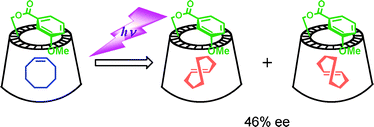 Supramolecular enantiodifferentiating photoisomerization of cyclooctene with modified β-cyclodextrins: critical control by a host structureRunhua Lu, Cheng Yang, Yujuan Cao, Zhizhong Wang, Takehiko Wada, Wei Jiao, Tadashi Mori, and Yoshihisa Inoue*Chem. Commun., 2008, 3, 374–376.
Supramolecular enantiodifferentiating photoisomerization of cyclooctene with modified β-cyclodextrins: critical control by a host structureRunhua Lu, Cheng Yang, Yujuan Cao, Zhizhong Wang, Takehiko Wada, Wei Jiao, Tadashi Mori, and Yoshihisa Inoue*Chem. Commun., 2008, 3, 374–376.Enantiodifferentiating photoisomerization of (Z)-cyclooctene included and sensitized by m-methoxybenzoyl-β-cyclodextrin gave chiral (E)-isomers in up to 46% enantiomeric excess, which is the highest value ever reported for supramolecular photochirogenesis with analogous hosts, thus demonstrating the crucial role of the sensitizer-spacer moiety in supramolecular photochirogenic systems.
@article{lu2008supramolecular, title = {Supramolecular enantiodifferentiating photoisomerization of cyclooctene with modified $\beta$-cyclodextrins: critical control by a host structure}, author = {Lu, Runhua and Yang, Cheng and Cao, Yujuan and Wang, Zhizhong and Wada, Takehiko and Jiao, Wei and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chem. Commun.}, volume = {3}, pages = {374--376}, year = {2008}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b714300a}, url = {https://doi.org/10.1039/b714300a}, dimensions = {true}, tab = {paper}, } -
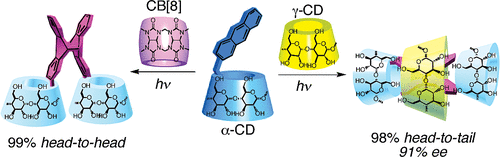 Highly stereoselective photocyclodimerization of α-cyclodextrin-appended anthracene mediated by γ-cyclodextrin and cucurbit [8] uril: a dramatic steric effect operating outside the binding siteCheng Yang, Tadashi Mori, Yumi Origane, Young Ho Ko, Narayanan Selvapalam, Kimoon Kim, and Yoshihisa Inoue*J. Am. Chem. Soc., 2008, 130, 8574–8575.
Highly stereoselective photocyclodimerization of α-cyclodextrin-appended anthracene mediated by γ-cyclodextrin and cucurbit [8] uril: a dramatic steric effect operating outside the binding siteCheng Yang, Tadashi Mori, Yumi Origane, Young Ho Ko, Narayanan Selvapalam, Kimoon Kim, and Yoshihisa Inoue*J. Am. Chem. Soc., 2008, 130, 8574–8575.Photocyclodimerization of α-cyclodextrin (CD)-appended anthracene was studied in the presence of γ-CD and cucurbit[8]uril (CB[8]) hosts to manipulate the stereodifferentiating photoreaction occurring inside the cavity by the bulky attachment located outside. The γ-CD-mediated photodimerization afforded the head-to-tail photodimers in 98% combined yield, in particular, the syn-head-to-tail photodimer of 91% ee in 68% yield, which are much greater than 32% ee and 44% yield obtained with unmodified anthracene carboxylate. The use of CB[8] also led to a striking inversion of the head-to-tail/head-to-head selectivity, affording exclusively the head-to-head photodimers in 99% combined yield.
@article{yang2008highly, title = {Highly stereoselective photocyclodimerization of α-cyclodextrin-appended anthracene mediated by γ-cyclodextrin and cucurbit [8] uril: a dramatic steric effect operating outside the binding site}, author = {Yang, Cheng and Mori, Tadashi and Origane, Yumi and Ko, Young Ho and Selvapalam, Narayanan and Kim, Kimoon and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {130}, number = {27}, pages = {8574--8575}, year = {2008}, publisher = {ACS Publications}, doi = {10.1021/ja8032923}, url = {https://doi.org/10.1021/ja8032923}, dimensions = {true}, tab = {paper}, } -
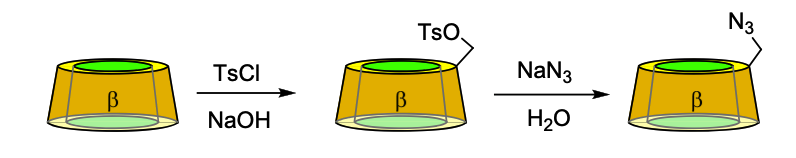 Synthesis of functionalized β-cyclodextrins by “click chemistry”Chenfeng Ke, Cheng Yang, Zixin Yang, Weijia Wu, Tadashi Mori*, Yoshihisa Inoue*, and Yu Liu*Heterocycles, 2008, 76, 155–160.
Synthesis of functionalized β-cyclodextrins by “click chemistry”Chenfeng Ke, Cheng Yang, Zixin Yang, Weijia Wu, Tadashi Mori*, Yoshihisa Inoue*, and Yu Liu*Heterocycles, 2008, 76, 155–160.Two new β-cyclodextrins (β-CDs) modified with chromophore were synthesized in high yields through Huisgen 1,3-dipolar cycloaddition. The amount of Cu catalyst was demonstrated to be a key factor that determines the yield of the 1,3-dipolar cycloaddition when applied to CD derivatization. While a catalytic amount of Cu-catalyst is commonly required in conventional click chemistry, more than a half equivalent of Cu catalyst was desirable for obtaining the modified CDs in satisfactory yields.
@article{ke2008synthesis, title = {Synthesis of functionalized β-cyclodextrins by “click chemistry”}, author = {Ke, Chenfeng and Yang, Cheng and Yang, Zixin and Wu, Weijia and Mori, Tadashi and Inoue, Yoshihisa and Liu, Yu}, journal = {Heterocycles}, volume = {76}, number = {1}, pages = {155--160}, year = {2008}, publisher = {Japan Institute of Heterocyclic Chemistry}, doi = {10.3987/COM-08-S(N)16}, url = {https://doi.org/10.3987/COM-08-S(N)16}, dimensions = {true}, tab = {paper}, } -
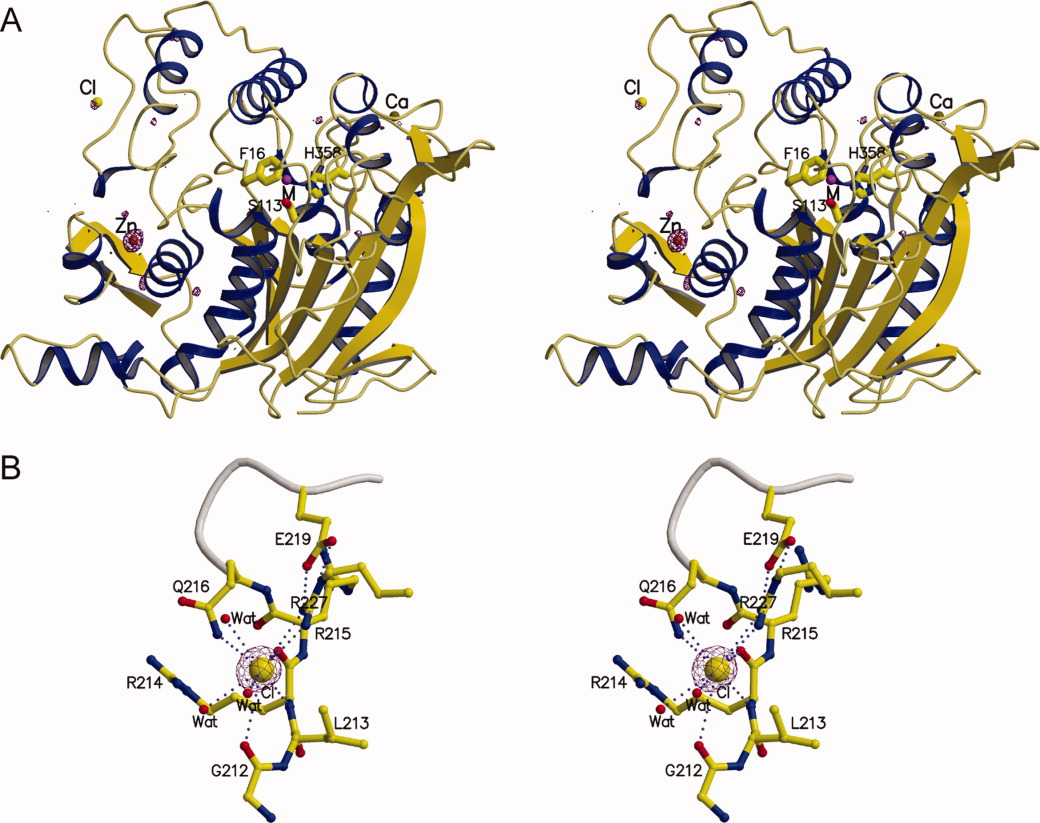 Novel cation-π interaction revealed by crystal structure of thermoalkalophilic lipaseHiroyoshi Matsumura*, Takahiko Yamamoto, Thean Chor Leow, Tadashi Mori, Abu Bakar Salleh, Mahiran Basri, Tsuyoshi Inoue, Yasushi Kai, and Raja Noor Zaliha Raja Abd Rahman*Proteins, 2008, 70, 592–598.
Novel cation-π interaction revealed by crystal structure of thermoalkalophilic lipaseHiroyoshi Matsumura*, Takahiko Yamamoto, Thean Chor Leow, Tadashi Mori, Abu Bakar Salleh, Mahiran Basri, Tsuyoshi Inoue, Yasushi Kai, and Raja Noor Zaliha Raja Abd Rahman*Proteins, 2008, 70, 592–598.Cation-π interactions are unique binding motifs that frequently occur between electron-rich aromatic ring and organic and inorganic (metallic) cations. This noncovalent interaction can be strong, as has been confirmed by the solid state studies of small molecule crystal structures,1, 2 and the theoretical and experimental analyses in aqueous media.3 A previous protein database search showed that the cation-π interaction can occur for every 77 amino acid residues in proteins, where the positively charged amino acids (e.g., arginine and lysine) and the aromatic amino acids (e.g., tryptophan, tyrosine, phenylananine) are usually involved.4 Cation-π interactions are therefore considered to be an essential force in generating the tertiary and quaternary structures of proteins that are induced by oligomerization and protein folding. These interactions are also important in biological processes such as protein–ligand1, 5, 6 and protein–DNA7-9 complex formations.
@article{matsumura2008novel, title = {Novel cation-π interaction revealed by crystal structure of thermoalkalophilic lipase}, author = {Matsumura, Hiroyoshi and Yamamoto, Takahiko and Leow, Thean Chor and Mori, Tadashi and Salleh, Abu Bakar and Basri, Mahiran and Inoue, Tsuyoshi and Kai, Yasushi and Rahman, Raja Noor Zaliha Raja Abd}, journal = {Proteins}, volume = {70}, number = {2}, pages = {592--598}, year = {2008}, publisher = {Wiley Online Library}, doi = {10.1002/prot.21799}, url = {https://doi.org/10.1002/prot.21799}, dimensions = {true}, tab = {paper}, }
2007
-
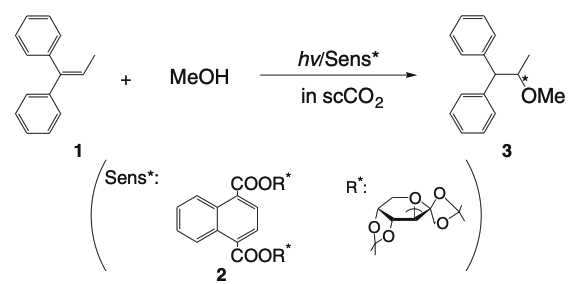 Critical control by temperature and pressure of enantiodifferentiating anti-Markovnikov photoaddition of methanol to diphenylpropene in near critical and supercritical carbon dioxideYasuhiro Nishiyama, Takehiko Wada, Tadashi Mori, and Yoshihisa Inoue*Chem. Lett., 2007, 36, 1488–1489.
Critical control by temperature and pressure of enantiodifferentiating anti-Markovnikov photoaddition of methanol to diphenylpropene in near critical and supercritical carbon dioxideYasuhiro Nishiyama, Takehiko Wada, Tadashi Mori, and Yoshihisa Inoue*Chem. Lett., 2007, 36, 1488–1489.The enantiomeric excess of photoadduct obtained in the title reaction was critically manipulated not only by pressure (P) but also by temperature (T), exhibiting substantially different P-dependence profiles at different T, yet sharing a sudden leap near the critical density at each T, for which the difference in clustering property of methanol in near and supercritical CO2 is thought to be responsible.
@article{nishiyama2007critical, title = {Critical control by temperature and pressure of enantiodifferentiating anti-Markovnikov photoaddition of methanol to diphenylpropene in near critical and supercritical carbon dioxide}, author = {Nishiyama, Yasuhiro and Wada, Takehiko and Mori, Tadashi and Inoue, Yoshihisa}, journal = {Chem. Lett.}, volume = {36}, number = {12}, pages = {1488--1489}, year = {2007}, publisher = {Oxford University Press}, doi = {10.1246/cl.2007.1488}, url = {https://doi.org/10.1246/cl.2007.1488}, dimensions = {true}, tab = {paper}, } -
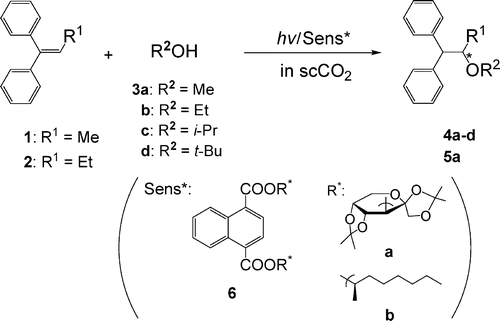 Mechanistic study on the enantiodifferentiating anti-Markovnikov photoaddition of alcohols to 1, 1-diphenyl-1-alkenes in near-critical and supercritical carbon dioxideYasuhiro Nishiyama, Masayuki Kaneda, Sadayuki Asaoka, Ryota Saito, Tadashi Mori, Takehiko Wada, and Inoue* YoshihisaJ. Phys. Chem. A, 2007, 111, 13432–13440.
Mechanistic study on the enantiodifferentiating anti-Markovnikov photoaddition of alcohols to 1, 1-diphenyl-1-alkenes in near-critical and supercritical carbon dioxideYasuhiro Nishiyama, Masayuki Kaneda, Sadayuki Asaoka, Ryota Saito, Tadashi Mori, Takehiko Wada, and Inoue* YoshihisaJ. Phys. Chem. A, 2007, 111, 13432–13440.Enantiodifferentiating anti-Markovnikov photoaddition of alcohol (methanol, ethanol, 2-propanol, and tert-butanol) to aromatic alkene (1,1-diphenylpropene and 1,1-diphenyl-1-butene), sensitized by optically active alkyl and saccharide naphthalene(di)carboxylates, was investigated in supercritical carbon dioxide at varying pressures to elucidate the effects of clustering on photosensitization and enantiodifferentiation behavior, in particular on the product’s enantiomeric excess (ee). For all the alkene/alcohol/chiral sensitizer combinations examined, a sudden change in the product’s ee was consistently observed near the critical density, which is attributable to the critical pressure dependence of clustering around the intervening exciplex intermediate.
@article{nishiyama2007mechanistic, title = {Mechanistic study on the enantiodifferentiating anti-Markovnikov photoaddition of alcohols to 1, 1-diphenyl-1-alkenes in near-critical and supercritical carbon dioxide}, author = {Nishiyama, Yasuhiro and Kaneda, Masayuki and Asaoka, Sadayuki and Saito, Ryota and Mori, Tadashi and Wada, Takehiko and Yoshihisa, Inoue}, journal = {J. Phys. Chem. A}, volume = {111}, number = {51}, pages = {13432--13440}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/jp076179i}, url = {https://doi.org/10.1021/jp076179i}, dimensions = {true}, tab = {paper}, } -
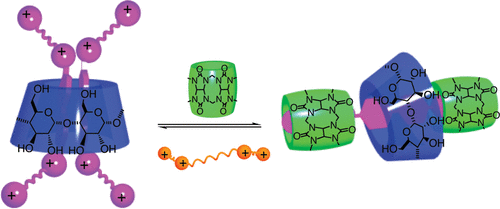 Dynamic switching between single-and double-axial rotaxanes manipulated by charge and bulkiness of axle terminiCheng Yang, Young Ho Ko, Narayanan Selvapalam, Yumi Origane, Tadashi Mori, Takehiko Wada, Kimoon Kim*, and Yoshihisa Inoue*Org. Lett., 2007, 9, 4789–4792.
Dynamic switching between single-and double-axial rotaxanes manipulated by charge and bulkiness of axle terminiCheng Yang, Young Ho Ko, Narayanan Selvapalam, Yumi Origane, Tadashi Mori, Takehiko Wada, Kimoon Kim*, and Yoshihisa Inoue*Org. Lett., 2007, 9, 4789–4792.Twin-axial [3]pseudorotaxanes, in which two multicharged axles simultaneously thread through the γ-CD cavity, were formed for the first time in solution. The twin-axial [3]pseudorotaxane was converted exclusively to a CB [6]-stoppered [4]pseudorotaxane by the addition of CB[6] but regenerated from the [4]pseudorotaxane by the addition of spermine, implementing an unprecedented switching of single/twin-axial rotaxanation.
@article{yang2007dynamic, title = {Dynamic switching between single-and double-axial rotaxanes manipulated by charge and bulkiness of axle termini}, author = {Yang, Cheng and Ko, Young Ho and Selvapalam, Narayanan and Origane, Yumi and Mori, Tadashi and Wada, Takehiko and Kim, Kimoon and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {9}, number = {23}, pages = {4789--4792}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/ol702142j}, url = {https://doi.org/10.1021/ol702142j}, dimensions = {true}, tab = {paper}, } -
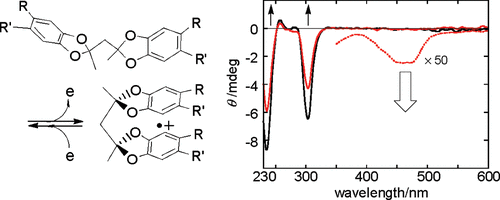 A new class of chiroptical molecular switches based on the redox-induced conformational changesMasato Fukui, Tadashi Mori*, Yoshihisa Inoue, and Rajendra Rathore*Org. Lett., 2007, 9, 3977–3980.
A new class of chiroptical molecular switches based on the redox-induced conformational changesMasato Fukui, Tadashi Mori*, Yoshihisa Inoue, and Rajendra Rathore*Org. Lett., 2007, 9, 3977–3980.A series of optically active bis(catecholketal)s 1−3 were prepared, and their chiroptical properties were investigated experimentally and theoretically, demonstrating that they undergo conformational changes upon 1-e- oxidation and can be used as redox-responsive chiroptical molecular switches.
@article{fukui2007new, title = {A new class of chiroptical molecular switches based on the redox-induced conformational changes}, author = {Fukui, Masato and Mori, Tadashi and Inoue, Yoshihisa and Rathore, Rajendra}, journal = {Org. Lett.}, volume = {9}, number = {20}, pages = {3977--3980}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/ol701639u}, url = {https://doi.org/10.1021/ol701639u}, dimensions = {true}, tab = {paper}, } -
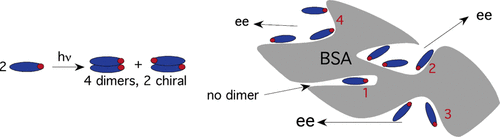 Supramolecular photochirogenesis with biomolecules. Mechanistic studies on the enantiodifferentiation for the photocyclodimerization of 2-anthracenecarboxylate mediated by bovine serum albuminMasaki Nishijima, Tamara CS Pace, Asao Nakamura, Tadashi Mori, Takehiko Wada, Cornelia Bohne*, and Yoshihisa Inoue*J. Org. Chem., 2007, 72, 2707–2715.
Supramolecular photochirogenesis with biomolecules. Mechanistic studies on the enantiodifferentiation for the photocyclodimerization of 2-anthracenecarboxylate mediated by bovine serum albuminMasaki Nishijima, Tamara CS Pace, Asao Nakamura, Tadashi Mori, Takehiko Wada, Cornelia Bohne*, and Yoshihisa Inoue*J. Org. Chem., 2007, 72, 2707–2715.Photophysics and photochemistry of 2-anthracenecarboxylate (AC) bound to bovine serum albumin (BSA) were investigated in detail for the first time by electronic absorption, circular dichroism (CD), steady-state and time-resolved fluorescence, fluorescence quenching, and product analysis studies. Through the spectroscopic investigations, it was revealed that the four independent binding pockets of BSA, which are known to accommodate 1, 3, 2, and 3 AC molecules in the order of decreasing affinity, are distinctly different in hydrophobicity, chiral environment, and accessibility. Interestingly, AC bound to site 1 gave highly structured fluorescence with dual lifetimes of 4.8 and 2.1 ns in an intensity ratio of 3:2, which may be assigned to the existence of two positional or orientational isomers within the very hydrophobic site 1. In contrast, the lifetime of AC in site 2 was much longer (13.3 ns), and ACs in sites 3 and 4 have broader fluorescence spectra with lifetimes that were practically indistinguishable from that in bulk water (15.8 ns). Although each of sites 2−4 simultaneously binds multiple AC molecules, no CD exciton coupling or static fluorescence quenching was detected, indicating that ACs bound to each site are not in close proximity to each other. Quenching studies with nitromethane further confirmed the significant difference in accessibility among the binding sites; thus, ACs bound to sites 1 and 2 are highly protected from the attack of the quencher, affording 32 and 10 times smaller rate constants than that for free AC in water. Product studies in the presence and absence of nitromethane more clearly revealed the photochirogenic performance of each binding site. Although the addition of nitromethane did not greatly alter the product distribution, the enantiomeric excesses (ee’s) of chiral cycloadducts 2 and 3 were critically manipulated by selectively retarding the photoreaction occurring at the more accessible binding sites. Thus, the highest ee of 38% was obtained for 2 in the presence of 18 mM nitromethane, while the highest ee of 58% was attained for 3 in the absence of nitromethane, both at [AC]/[BSA] = 3.6.
@article{nishijima2007supramolecular, title = {Supramolecular photochirogenesis with biomolecules. Mechanistic studies on the enantiodifferentiation for the photocyclodimerization of 2-anthracenecarboxylate mediated by bovine serum albumin}, author = {Nishijima, Masaki and Pace, Tamara CS and Nakamura, Asao and Mori, Tadashi and Wada, Takehiko and Bohne, Cornelia and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {72}, number = {8}, pages = {2707--2715}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/jo062226b}, url = {https://doi.org/10.1021/jo062226b}, dimensions = {true}, tab = {paper}, } -
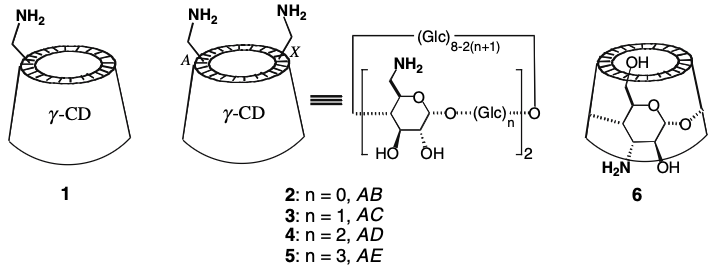 Enhanced ternary 1: 2 host–guest complexation of amino-γ-cyclodextrins with 2-anthracenecarboxylic acidCheng Yang, Gaku Fukuhara, Asao Nakamura, Yumi Origane, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Incl. Phenom. Macrocycl. Chem., 2007, 57, 433–437.
Enhanced ternary 1: 2 host–guest complexation of amino-γ-cyclodextrins with 2-anthracenecarboxylic acidCheng Yang, Gaku Fukuhara, Asao Nakamura, Yumi Origane, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Incl. Phenom. Macrocycl. Chem., 2007, 57, 433–437.The complexation behavior of 6-amino-6-deoxy-γ-cyclodextrin (CD), 6A,6X-diamino-6A,6X-deoxy-γ-CDs and 3A-amino-3A-deoxy-altro-γ-CD with 2-anthracenecarboxylic acid (AC) was studied by NMR, UV–vis and circular dichroism spectroscopy. These modified γ-CD derivatives were found to form stable 1:2 host-guest ternary complexes with AC in aqueous solution. Compared with native γ-CD, the primary-face-aminated γ-CDs exhibited remarkably enhanced overall association constants as a result of the additional electrostatic interactions between the oppositely charged host and guest. In contrast, the ternary complex formation of the secondary-face-aminated γ-CD with AC was hindered.
@article{yang2007enhanced, title = {Enhanced ternary 1: 2 host--guest complexation of amino-$\gamma$-cyclodextrins with 2-anthracenecarboxylic acid}, author = {Yang, Cheng and Fukuhara, Gaku and Nakamura, Asao and Origane, Yumi and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {J. Incl. Phenom. Macrocycl. Chem.}, volume = {57}, number = {1}, pages = {433--437}, year = {2007}, publisher = {Springer}, doi = {10.1007/s10847-006-9230-y}, url = {https://doi.org/10.1007/s10847-006-9230-y}, dimensions = {true}, tab = {paper}, } -
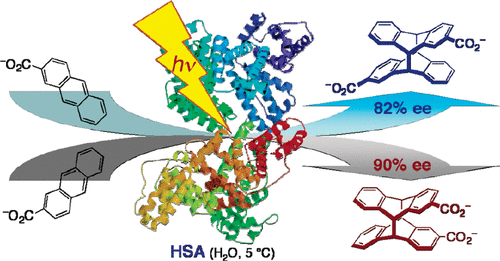 Highly enantiomeric supramolecular [4+ 4] photocyclodimerization of 2-anthracenecarboxylate mediated by human serum albuminMasaki Nishijima, Takehiko Wada, Tadashi Mori, Tamara CS Pace, Cornelia Bohne*, and Yoshihisa Inoue*J. Am. Chem. Soc., 2007, 129, 3478–3479.
Highly enantiomeric supramolecular [4+ 4] photocyclodimerization of 2-anthracenecarboxylate mediated by human serum albuminMasaki Nishijima, Takehiko Wada, Tadashi Mori, Tamara CS Pace, Cornelia Bohne*, and Yoshihisa Inoue*J. Am. Chem. Soc., 2007, 129, 3478–3479.Photoirradiation at λ > 320 nm of 2-anthracenecarboxylate bound to human serum albumin in an aqueous buffer solution at 5 °C gave syn head-to-tail cyclodimer 2 in 82% ee and anti head-to-head cyclodimer 3 in 90% ee.
@article{nishijima2007highly, title = {Highly enantiomeric supramolecular [4+ 4] photocyclodimerization of 2-anthracenecarboxylate mediated by human serum albumin}, author = {Nishijima, Masaki and Wada, Takehiko and Mori, Tadashi and Pace, Tamara CS and Bohne, Cornelia and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {129}, number = {12}, pages = {3478--3479}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/ja068475z}, url = {https://doi.org/10.1021/ja068475z}, dimensions = {true}, tab = {paper}, } -
 Inherently chiral molecular clips: synthesis, chiroptical properties, and application to chiral discriminationGaku Fukuhara, Süreyya Madenci, Jolanta Polkowska, Frank Bastkowski, Frank-Gerrit Klärner, Yumi Origane*, Masayuki Kaneda, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Eur. J., 2007, 13, 2473–2479.
Inherently chiral molecular clips: synthesis, chiroptical properties, and application to chiral discriminationGaku Fukuhara, Süreyya Madenci, Jolanta Polkowska, Frank Bastkowski, Frank-Gerrit Klärner, Yumi Origane*, Masayuki Kaneda, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Eur. J., 2007, 13, 2473–2479.Inherently chiral molecular clips (MCs), pseudoenantiomeric anti-1 and anti-2, as well as mesoid syn-3, were synthesized by diastereodifferentiating repetitive Diels–Alder reactions of the achiral bisdienophile 6 with chiral diene 5 generated in situ from (−)-menthyl 3,4-bis(dibromomethyl)benzoate 4. These MCs were successfully separated by chiral HPLC to give optically active anti-1 and anti-2 and almost optically inactive syn-3. The structures of anti-1, anti-2, and syn-3 were assigned by high-resolution NMR and the absolute configurations of anti-1 and anti-2 were determined by the exciton-chirality method. Optically active anti-2 can serve as a chiral host. It binds the HCl adduct of D-tryptophan methyl ester (D-TrpOMe⋅HCl) 3.5 times stronger than the L-enantiomer (KD/KL=3.5).
@article{fukuhara2007inherently, title = {Inherently chiral molecular clips: synthesis, chiroptical properties, and application to chiral discrimination}, author = {Fukuhara, Gaku and Madenci, Süreyya and Polkowska, Jolanta and Bastkowski, Frank and Klärner, Frank-Gerrit and Origane, Yumi and Kaneda, Masayuki and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Eur. J.}, volume = {13}, number = {9}, pages = {2473--2479}, year = {2007}, publisher = {Wiley Online Library}, doi = {10.1002/chem.200601585}, url = {https://doi.org/10.1002/chem.200601585}, dimensions = {true}, tab = {paper}, } -
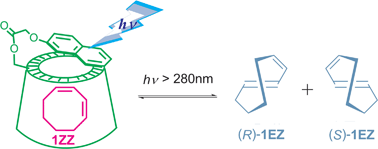 Supramolecular enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene included and sensitized by naphthalene-modified cyclodextrinsCheng Yang, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*New J. Chem., 2007, 31, 697–702.
Supramolecular enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene included and sensitized by naphthalene-modified cyclodextrinsCheng Yang, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*New J. Chem., 2007, 31, 697–702.Three naphthalene-modified cyclodextrins (CDs) 3–5 were synthesized as supramolecular chiral photosensitizing hosts for enantiodifferentiating photoisomerization of (Z,Z)-1,3-cyclooctadiene (1ZZ) to its E,Z-isomer (1EZ). In aqueous methanolic solutions, β-CD-based sensitizer 4 binds 1ZZ in its chiral cavity much more strongly than α- and γ-CD homologues 3 and 5. Accelerated, often static, fluorescence quenching of these naphthalene-modified CDs was observed upon inclusion of 1ZZ. The photoisomerization of 1ZZ mediated by 3–5 yielded enantiomeric 1EZ in modest yields. The enantiomeric excesses (ee’s) obtained with α- and β-CD-based sensitizers 3 and 4, both of which have relatively small cavities, are less sensitive to temperature, demonstrating the low-entropy nature of the α- and β-CD complexes. In contrast, increasing reaction temperature significantly diminished the product’s ee and even caused a switching of enantioselectivity upon photoisomerization sensitized by γ-CD-based 5, revealing that the entropy factor plays a crucial role in the wide cavity of γ-CD.
@article{yang2007supramolecular, title = {Supramolecular enantiodifferentiating photoisomerization of (Z, Z)-1, 3-cyclooctadiene included and sensitized by naphthalene-modified cyclodextrins}, author = {Yang, Cheng and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {New J. Chem.}, volume = {31}, number = {5}, pages = {697--702}, year = {2007}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b615353d}, url = {https://doi.org/10.1039/b615353d}, dimensions = {true}, tab = {paper}, } -
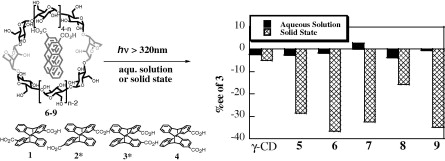 A remarkable stereoselectivity switching upon solid-state versus solution-phase enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid mediated by native and 3, 6-anhydro-γ-cyclodextrinsCheng Yang*, Masaki Nishijima, Asao Nakamura, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Tetrahedron Lett., 2007, 48, 4357–4360.
A remarkable stereoselectivity switching upon solid-state versus solution-phase enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid mediated by native and 3, 6-anhydro-γ-cyclodextrinsCheng Yang*, Masaki Nishijima, Asao Nakamura, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Tetrahedron Lett., 2007, 48, 4357–4360.The enantiodifferentiating [4+4] photocyclodimerization of anthracenecarboxylic acid (AC) mediated by native, mono- and di-3,6-anhydro-γ-cyclodextrins was investigated in both aqueous solution and solid-state. The solid-state photolyses gave inherently disfavored head-to-head photodimers in much higher chemical and optical yields than in the aqueous solution.
@article{yang2007remarkable, title = {A remarkable stereoselectivity switching upon solid-state versus solution-phase enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylic acid mediated by native and 3, 6-anhydro-γ-cyclodextrins}, author = {Yang, Cheng and Nishijima, Masaki and Nakamura, Asao and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Tetrahedron Lett.}, volume = {48}, number = {25}, pages = {4357--4360}, year = {2007}, publisher = {Elsevier}, doi = {10.1016/j.tetlet.2007.04.104}, url = {https://doi.org/10.1016/j.tetlet.2007.04.104}, dimensions = {true}, tab = {paper}, } -
 A synthetic host-guest system achieves avidin-biotin affinity by overcoming enthalpy–entropy compensationMikhail V Rekharsky, Tadashi Mori, Cheng Yang, Young Ho Ko, N Selvapalam, Hyunuk Kim, David Sobransingh, Angel E Kaifer*, Simin Liu, Lyle Isaacs*, Wei Chen, Sarvin Moghaddam, Michael K Gilson*, Kimoon Kim*, and Yoshihisa Inoue*Proc. Natl. Acad. Sci., 2007, 104, 20737–20742.
A synthetic host-guest system achieves avidin-biotin affinity by overcoming enthalpy–entropy compensationMikhail V Rekharsky, Tadashi Mori, Cheng Yang, Young Ho Ko, N Selvapalam, Hyunuk Kim, David Sobransingh, Angel E Kaifer*, Simin Liu, Lyle Isaacs*, Wei Chen, Sarvin Moghaddam, Michael K Gilson*, Kimoon Kim*, and Yoshihisa Inoue*Proc. Natl. Acad. Sci., 2007, 104, 20737–20742.The molecular host cucurbit[7]uril forms an extremely stable inclusion complex with the dicationic ferrocene derivative bis(trimethylammoniomethyl)ferrocene in aqueous solution. The equilibrium association constant for this host-guest pair is 3 × 1015 M−1 (Kd = 3 × 10−16 M), equivalent to that exhibited by the avidin–biotin pair. Although purely synthetic systems with larger association constants have been reported, the present one is unique because it does not rely on polyvalency. Instead, it achieves its extreme affinity by overcoming the compensatory enthalpy–entropy relationship usually observed in supramolecular complexes. Its disproportionately low entropic cost is traced to extensive host desolvation and to the rigidity of both the host and the guest.
@article{rekharsky2007synthetic, title = {A synthetic host-guest system achieves avidin-biotin affinity by overcoming enthalpy--entropy compensation}, author = {Rekharsky, Mikhail V and Mori, Tadashi and Yang, Cheng and Ko, Young Ho and Selvapalam, N and Kim, Hyunuk and Sobransingh, David and Kaifer, Angel E and Liu, Simin and Isaacs, Lyle and Chen, Wei and Moghaddam, Sarvin and Gilson, Michael K and Kim, Kimoon and Inoue, Yoshihisa}, journal = {Proc. Natl. Acad. Sci.}, volume = {104}, number = {52}, pages = {20737--20742}, year = {2007}, publisher = {National Academy of Sciences}, doi = {10.1073/pnas.0706407105}, url = {https://doi.org/10.1073/pnas.0706407105}, dimensions = {true}, tab = {paper}, } -
 Quantum chemical study on the circular dichroism spectra and specific rotation of donor- acceptor cyclophanesTadashi Mori*, Yoshihisa Inoue, and Stefan Grimme*J. Phys. Chem. A, 2007, 111, 7995–8006.
Quantum chemical study on the circular dichroism spectra and specific rotation of donor- acceptor cyclophanesTadashi Mori*, Yoshihisa Inoue, and Stefan Grimme*J. Phys. Chem. A, 2007, 111, 7995–8006.The structures of donor,acceptor-substituted cyclophanes were optimized by DFT and MP2 methods and compared with the X-ray crystallographic structures. The electronic circular dichroism (CD) spectra of these chiral cyclophanes were simulated by time dependent density functional theory (TD−DFT) with several functionals including different amounts of “exact” Hartree−Fock exchange. The experimental oscillator and rotatory strengths were best reproduced by the BH-LYP/TZV2P method. The specific rotation and vibrational circular dichroism (VCD) spectra were also calculated at the BH-LYP/aug-cc-pVDZ and B3-LYP/6-31G(d) levels, respectively, and compared with the experimental data. Better performance was obtained with the ECD, rather than the specific rotation or the VCD spectral calculations in view of the computation time and accuracy for the determination of absolute configuration (AC). The exciton coupling model can be applied only for the cyclophanes without CT-character. However, the split pattern found in the experiment does not appear to originate from a simple two-transition coupling, indicating that this method should be applied with caution to the AC determination. This conclusion was supported by the TD−DFT investigations of the transition moments and the roles of excited-state electronic configuration associated with these split bands. Cyclophanes with donor−acceptor interactions showed Cotton effects at the CT band and couplets at the 1La and 1Lb bands. Although the degree of charge transfer between the rings is very small, as revealed by a Mulliken−Hash analysis, the split Cotton effects are due to a large separation in energy of the donor and acceptor orbitals. The effect of the distance and angle between the donor and acceptor moieties in model (intermolecular) CT complexes on the calculated CD spectra was also studied and compared with those obtained for various paracyclophanes.
@article{mori2007quantum, title = {Quantum chemical study on the circular dichroism spectra and specific rotation of donor- acceptor cyclophanes}, author = {Mori, Tadashi and Inoue, Yoshihisa and Grimme, Stefan}, journal = {J. Phys. Chem. A}, volume = {111}, number = {32}, pages = {7995--8006}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/jp073596m}, url = {https://doi.org/10.1021/jp073596m}, dimensions = {true}, tab = {paper}, } -
 Experimental and Theoretical Study of the CD Spectra and Conformational Properties of Axially Chiral 2, 2 ‘-, 3, 3 ‘-, and 4, 4 ‘-Biphenol EthersTadashi Mori*, Yoshihisa Inoue, and Stefan GrimmeJ. Phys. Chem. A, 2007, 111, 4222–4234.
Experimental and Theoretical Study of the CD Spectra and Conformational Properties of Axially Chiral 2, 2 ‘-, 3, 3 ‘-, and 4, 4 ‘-Biphenol EthersTadashi Mori*, Yoshihisa Inoue, and Stefan GrimmeJ. Phys. Chem. A, 2007, 111, 4222–4234.@article{mori2007experimental, title = {Experimental and Theoretical Study of the CD Spectra and Conformational Properties of Axially Chiral 2, 2 ‘-, 3, 3 ‘-, and 4, 4 ‘-Biphenol Ethers}, author = {Mori, Tadashi and Inoue, Yoshihisa and Grimme, Stefan}, journal = {J. Phys. Chem. A}, volume = {111}, number = {20}, pages = {4222--4234}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/jp071709w}, url = {https://doi.org/10.1021/jp071709w}, dimensions = {true}, tab = {paper}, } -
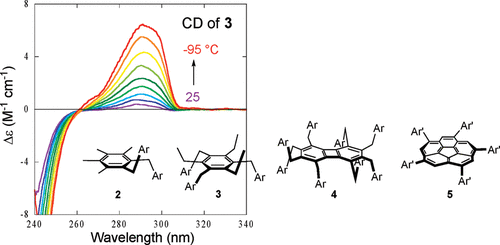 A combined experimental and theoretical study on the conformation of multiarmed chiral aryl ethersTadashi Mori*, Stefan Grimme, and Yoshihisa InoueJ. Org. Chem., 2007, 72, 6998–7010.
A combined experimental and theoretical study on the conformation of multiarmed chiral aryl ethersTadashi Mori*, Stefan Grimme, and Yoshihisa InoueJ. Org. Chem., 2007, 72, 6998–7010.Four series of multiarmed chiral aryl ethers carrying two, three, five, or eight side-chains on a variety of aromatic core molecules (2−5) were prepared. The structure and conformation of 2 and 3 (in the solid state) were determined by the X-ray crystallographic analyses. While a pair of alternated (anti) conformers (i.e, up−down and down−up) were found in the crystal of 2, three side-arms in 3 were aligned in the same direction to give a C3-symmetric syn-conformation. Examinations by dispersion-corrected density functional (DFT-D) calculations revealed that two out of six anti- and two out of four syn-conformers of 2 are energetically most important. Two calculated structures of anti-conformers are in good agreement with those found in the solid state by X-ray analysis. Similarly, relevant conformations of syn-3, fully alternated 4, and C5-symmetric 5 were optimized at the DFT-D-B-LYP/TZVP level. The structure and conformation of the side-arms in 2−5 in solution were further studied by temperature dependent 1H NMR and UV−vis spectroscopy. In addition, comparative experimental and theoretical CD spectral studies were carried out in order to elucidate the contribution of the thermodynamically less-stable minor isomers in solution. The CD spectral changes observed for 2 and 3 at varying temperatures were quite different, while the parent chiral arene 1, as well as 4 and 5, only showed an increased intensity of the negative Cotton effect for the 1Lb band. The latter behavior is readily accounted for in terms of the conformational freezing of the chiral groups at low temperatures. The unusual CD spectral behavior observed for 2 and 3 was rationalized by the conformational alteration of the side-arms. Because of attractive van der Waals interactions between the aromatic units of the arms in nonpolar solvents, the syn-conformations become gradually more important for 2 at low temperatures, which eventually results in a weak positive Cotton effect for the 1Lb band. This was also supported by the SCS-MP2/TZVPP single-point energy calculations for the relevant conformers of 2. For 3, the contribution of the C3-symmetrical conformer becomes more important than the less-symmetrical isomers at low temperatures. The conformations of 2 and 3 in their excited states as well as in the oxidized states were also examined.
@article{mori2007combined, title = {A combined experimental and theoretical study on the conformation of multiarmed chiral aryl ethers}, author = {Mori, Tadashi and Grimme, Stefan and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {72}, number = {18}, pages = {6998--7010}, year = {2007}, publisher = {ACS Publications}, doi = {10.1021/jo071216n}, url = {https://doi.org/10.1021/jo071216n}, dimensions = {true}, tab = {paper}, }
2006
-
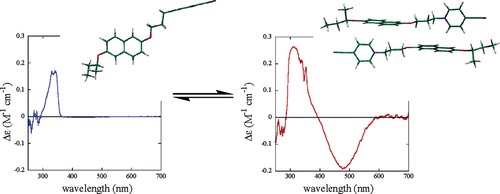 Time dependent density functional theory calculations for electronic circular dichroism spectra and optical rotations of conformationally flexible chiral donor- acceptor dyadTadashi Mori*, Yoshihisa Inoue, and Stefan Grimme*J. Org. Chem., 2006, 71, 9797–9806.
Time dependent density functional theory calculations for electronic circular dichroism spectra and optical rotations of conformationally flexible chiral donor- acceptor dyadTadashi Mori*, Yoshihisa Inoue, and Stefan Grimme*J. Org. Chem., 2006, 71, 9797–9806.Twelve conformations of a chiral donor−acceptor (charge-transfer) dyad and six conformations of its dimer complex were structurally optimized by using the Kohn−Sham density functional theory (BLYP/TZV2P) incorporating a recently developed empirical correction scheme that uses C6/R6 potentials for van der Waals interactions (DFT-D). Subsequent time-dependent DFT calculations with BH-LYP and B3-LYP functionals (with triple-ζ basis set) were performed to obtain theoretical circular dichroism (CD) spectra. The experimental CD spectra obtained independently were properly reproduced by averaging the calculated spectra of individual conformers according to a Boltzmann population derived from single-point SCS-MP2 energies. The optical rotations of the monomer were also calculated by using the same functionals with an aug-cc-pVDZ basis set. Dielectric continuum solvation models (COSMO) applied to correct the relative energies from the isolated molecule calculations resulted in conformer distributions that piled the same or even poorer level of agreement with the experimental CD spectrum. Our results clearly show the advantage of the DFT-D method for the geometry optimization of large systems with donor−acceptor interactions and the TD-DFT/BH-LYP calculations for reproducing the experimental CD spectra. As compared with the calculated optical rotations, the wealthy information embedded in the experimental/calculated CD spectra is requisite for the configurational and/or conformational analyses of relatively large and flexible chiral organic molecules in solution.
@article{mori2006time, title = {Time dependent density functional theory calculations for electronic circular dichroism spectra and optical rotations of conformationally flexible chiral donor- acceptor dyad}, author = {Mori, Tadashi and Inoue, Yoshihisa and Grimme, Stefan}, journal = {J. Org. Chem.}, volume = {71}, number = {26}, pages = {9797--9806}, year = {2006}, publisher = {ACS Publications}, doi = {10.1021/jo061855i}, url = {https://doi.org/10.1021/jo061855i}, dimensions = {true}, tab = {paper}, } -
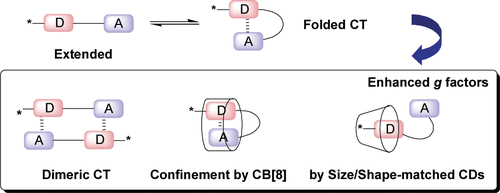 Circular dichroism of intra-and intermolecular charge-transfer complexes. Enhancement of anisotropy factors by dimer formation and by confinementTadashi Mori*, Young Ho Ko, Kimoon Kim, and Yoshihisa Inoue*J. Org. Chem., 2006, 71, 3232–3247.
Circular dichroism of intra-and intermolecular charge-transfer complexes. Enhancement of anisotropy factors by dimer formation and by confinementTadashi Mori*, Young Ho Ko, Kimoon Kim, and Yoshihisa Inoue*J. Org. Chem., 2006, 71, 3232–3247.The dynamic behavior of new CT-dyads has been studied by means of UV−vis, fluorescence, and NMR spectroscopies under a variety of conditions. It was found that the CT-dyads exhibit conformational variations, such as extended and folded monomers and an antiparallel dimer complex, depending on the conditions. The CT interaction was found in the folded conformation at ambient temperature, while the contribution of the dimeric species became evident at lower temperatures. Most interestingly, close examinations of the circular dichroism spectra of these CT-dyads reveal that the anisotropy (g) factors of the dimers are significantly enhanced by a factor of ∼30 in the CT transition region. Such enhancement is rationalized in terms of the stronger CT interactions in the dimer through the double electronic coupling element, which imposes stronger restrictions on the rotation of alkyl group(s). Confinement of the CT-dyads in cyclodextrin (CD) and cucurbituril cavities afforded further insights into the chiroptical properties of the CT-dyad. The effects of confinement are clearly size-dependent, exhibiting a substantial enhancement of the g factors by a factor of 5−10 upon inclusion by β-CD and also by cucurbit[8]uril, but with no appreciable changes upon complexation with the other CDs. These results indicate that the conformational fixation of CT-dyads, for example by dimer formation or by confinement in size/shape-matched cavities, is a conventional, yet powerful, tool for manipulating (mostly enhancing) the chiroptical properties of the CT transition, which should be applicable in general to a variety of molecular and supramolecular CT systems.
@article{mori2006circular, title = {Circular dichroism of intra-and intermolecular charge-transfer complexes. Enhancement of anisotropy factors by dimer formation and by confinement}, author = {Mori, Tadashi and Ko, Young Ho and Kim, Kimoon and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {71}, number = {8}, pages = {3232--3247}, year = {2006}, publisher = {ACS Publications}, doi = {10.1021/jo0602672}, url = {https://doi.org/10.1021/jo0602672}, dimensions = {true}, tab = {paper}, } -
 Switching of product’s chirality in diastereodifferentiating [2+ 2] photocycloaddition of (E)-versus (Z)-stilbene to chiral fumarate upon direct and charge-transfer-band excitationHideaki Saito, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*Org. Lett., 2006, 8, 1909–1912.
Switching of product’s chirality in diastereodifferentiating [2+ 2] photocycloaddition of (E)-versus (Z)-stilbene to chiral fumarate upon direct and charge-transfer-band excitationHideaki Saito, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*Org. Lett., 2006, 8, 1909–1912.Diastereodifferentiating [2+2] photocycloadditions of (E)- and (Z)-stilbenes to bis((R)-1-methylpropyl) fumarate were performed through the direct excitation of stilbenes and the selective excitation of the charge-transfer (CT) complex at various temperatures. The geometrical isomers of stilbene afforded the opposite diastereomers of μ-truxinate in both excitation modes, with a dramatic decrease in the product’s diastereoselectivity upon prolonged irradiations.
@article{saito2006switching, title = {Switching of product's chirality in diastereodifferentiating [2+ 2] photocycloaddition of (E)-versus (Z)-stilbene to chiral fumarate upon direct and charge-transfer-band excitation}, author = {Saito, Hideaki and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {8}, number = {9}, pages = {1909--1912}, year = {2006}, publisher = {ACS Publications}, doi = {10.1021/ol060468s}, url = {https://doi.org/10.1021/ol060468s}, dimensions = {true}, tab = {paper}, } -
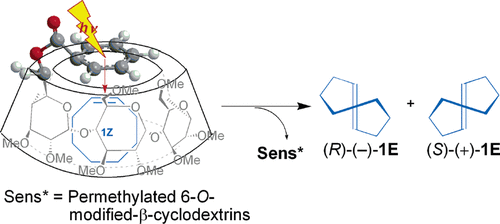 Entropy-Controlled Supramolecular Photochirogenesis: Enantiodifferentiating Z- E Photoisomerization of Cyclooctene Included and Sensitized by Permethylated 6-O-Modified β-CyclodextrinsGaku Fukuhara, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Org. Chem., 2006, 71, 8233–8243.
Entropy-Controlled Supramolecular Photochirogenesis: Enantiodifferentiating Z- E Photoisomerization of Cyclooctene Included and Sensitized by Permethylated 6-O-Modified β-CyclodextrinsGaku Fukuhara, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Org. Chem., 2006, 71, 8233–8243.Permethylated 6-O-modified β-cyclodextrins 2a−2d were synthesized as novel photosensitizing hosts with a flexible skeleton. Circular dichroism (CD) and 2D NMR spectral examinations of benzoate 2a revealed that the benzoate moiety is deeply included into its own cavity in aqueous solution. Upon addition of (Z)-cyclooctene (1Z) to a 50% aqueous methanol solution of 2a at 25 °C, the benzoate moiety of 2a was gradually excluded from the cavity as indicated by the CD spectral changes; the Job’s plot revealed the formation of a 1:1 complex of 2a with 1Z. The binding constants for the complexation of 1Z by 2a were determined by CD spectral titration in 50% aqueous methanol at various temperatures. The van’t Hoff analysis of the obtained data afforded the thermodynamic parameters (ΔH° = −3.1 kJ mol-1, ΔS° = 48.5 J mol-1 K-1), demonstrating the entropy-driven complexation by the permethylated cyclodextrin. This is in sharp contrast to the complexation of 1Z by nonmethylated β-cyclodextrin benzoate that is driven by enthalpy (ΔH° = −31.8 kJ mol-1 and ΔS° = −51.1 J mol-1 K-1). Upon supramolecular photosensitization with 2a−2d, 1Z isomerized to the (E)-isomer (1E) in moderate enantiomeric excesses (ee’s), which however displayed significant temperature dependence with accompanying switching of the product’s chirality in an extreme case. Such dynamic behavior of ee is very different from that reported for the photosensitization with nonmethylated cyclodextrin benzoate, where the product’s ee is controlled by host occupancy. Eyring treatment of the ee obtained at various temperatures (<0 °C) gave the differential activation parameters for the enantiodifferentiation process occurring in the supramolecular exciplex, revealing the crucial role of entropy, as indicated by the ΔΔS⧧ value changing dynamically from +4 to −24 J K-1 mol-1. The origin of the contrasting behavior of permethylated versus nonmethylated cyclodextrin hosts is inferred to be the conformational flexibility of the former host, which enables the entropy-driven guest complexation in the ground state and the entropy-controlled enantiodifferentiation in the excited state.
@article{fukuhara2006entropy, title = {Entropy-Controlled Supramolecular Photochirogenesis: Enantiodifferentiating Z- E Photoisomerization of Cyclooctene Included and Sensitized by Permethylated 6-O-Modified β-Cyclodextrins}, author = {Fukuhara, Gaku and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {J. Org. Chem.}, volume = {71}, number = {21}, pages = {8233--8243}, year = {2006}, publisher = {ACS Publications}, doi = {10.1021/jo061389x}, url = {https://doi.org/10.1021/jo061389x}, dimensions = {true}, tab = {paper}, } -
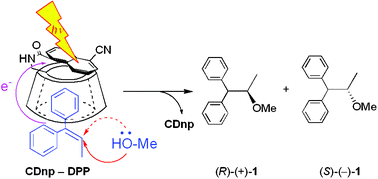 The first supramolecular photosensitization of enantiodifferentiating bimolecular reaction: anti-Markovnikov photoaddition of methanol to 1, 1-diphenylpropene sensitized by modified β-cyclodextrinGaku Fukuhara, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2006, 71, 1712–1714.
The first supramolecular photosensitization of enantiodifferentiating bimolecular reaction: anti-Markovnikov photoaddition of methanol to 1, 1-diphenylpropene sensitized by modified β-cyclodextrinGaku Fukuhara, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2006, 71, 1712–1714.Enantiodifferentiating polar photoaddition of methanol to 1,1-diphenylpropene included and sensitized by cyanonaphthalene-modified β-cyclodextrin was examined for the first time to give optically active anti-Markovnikov adduct with accompanying inversion of the chiral sense of the photoproduct by temperature, which is entropic in origin.
@article{fukuhara2006first, title = {The first supramolecular photosensitization of enantiodifferentiating bimolecular reaction: anti-Markovnikov photoaddition of methanol to 1, 1-diphenylpropene sensitized by modified β-cyclodextrin}, author = {Fukuhara, Gaku and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Commun.}, number = {16}, pages = {1712--1714}, year = {2006}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b601674j}, url = {https://doi.org/10.1039/b601674j}, dimensions = {true}, tab = {paper}, }
2005
-
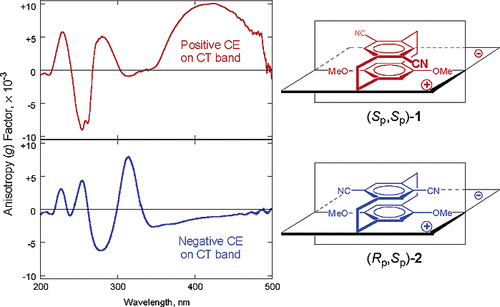 Absolute Configuration of Chiral [2.2]Paracyclophanes with Intramolecular Charge-Transfer Interaction. Failure of the Exciton Chirality Method and Use of the Sector Rule Applied to the Cotton Effect of the CT TransitionTakahiro Furo, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*J. Am. Chem. Soc., 2005, 127, 8242–8243.
Absolute Configuration of Chiral [2.2]Paracyclophanes with Intramolecular Charge-Transfer Interaction. Failure of the Exciton Chirality Method and Use of the Sector Rule Applied to the Cotton Effect of the CT TransitionTakahiro Furo, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*J. Am. Chem. Soc., 2005, 127, 8242–8243.Optically active 4,7-dicyano-12,15-dimethoxy[2.2]paracyclophanes have been separated by chiral HPLC and their absolute configurations determined by comparison of the experimental and the theoretical VCD spectra. X-ray crystallographic structures for both diastereomers are also reported. The electronic circular dichroism spectra of these enantiomeric pairs, as chiral intramolecular charge-transfer complexes, have been obtained for the first time. The exciton coupling method, usually used for determining the absolute configuration of chiral molecules, however, did not give a correct prediction for the present CT−paracyclophane system. Instead, empirical sector rules for the signs of the Cotton effects of the CT transition can be applied for the assignment of the absolute configuration.
@article{furo2005absolute, title = {Absolute Configuration of Chiral [2.2]Paracyclophanes with Intramolecular Charge-Transfer Interaction. Failure of the Exciton Chirality Method and Use of the Sector Rule Applied to the Cotton Effect of the CT Transition}, author = {Furo, Takahiro and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {127}, number = {23}, pages = {8242--8243}, year = {2005}, publisher = {ACS Publications}, doi = {10.1021/ja0508323}, url = {https://doi.org/10.1021/ja0508323}, dimensions = {true}, tab = {paper}, } -
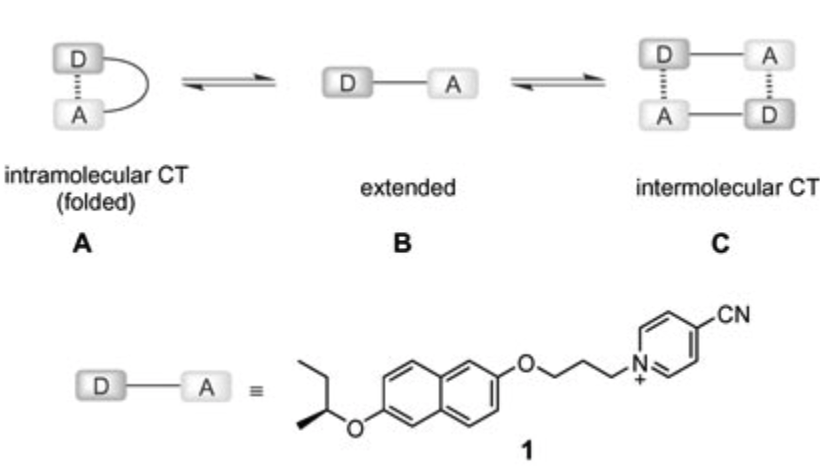 Circular Dichroism of a Chiral Tethered Donor-Acceptor System: Enhanced Anisotropy Factors in Charge-Transfer Transitions by Dimer Formation and by ConfinementTadashi Mori* and Yoshihisa Inoue*Angew. Chem. Int. Ed., 2005, 44, 2582–2585.
Circular Dichroism of a Chiral Tethered Donor-Acceptor System: Enhanced Anisotropy Factors in Charge-Transfer Transitions by Dimer Formation and by ConfinementTadashi Mori* and Yoshihisa Inoue*Angew. Chem. Int. Ed., 2005, 44, 2582–2585.A charge-transfer (CT) dyad with a trimethylene tether forms a folded or extended monomer or a dimer depending on the conditions (see picture). Both dimer formation at low temperatures and monomer confinement by inclusion in a cyclodextrin cavity greatly enhance the anisotropy factor of the CT transition of the complex.
@article{mori2005circular, title = {Circular Dichroism of a Chiral Tethered Donor-Acceptor System: Enhanced Anisotropy Factors in Charge-Transfer Transitions by Dimer Formation and by Confinement}, author = {Mori, Tadashi and Inoue, Yoshihisa}, journal = {Angew. Chem. Int. Ed.}, volume = {44}, number = {17}, pages = {2582--2585}, year = {2005}, publisher = {Weily}, doi = {10.1002/anie.200462071}, url = {https://doi.org/10.1002/anie.200462071}, dimensions = {true}, tab = {paper}, } -
 Enantiodifferentiating [4+ 4] photocyclodimerization of 2-anthracenecarboxylate catalyzed by 6A, 6X-diamino-6A, 6X-dideoxy-h-cyclodextrins: Manipulation of product chirality by electrostatic interaction, temperature and solvent in supramolecular photochirogenesisCheng Yang, Gaku Fukuhara, Asao Nakamura, Yumi Origane, Kahee Fujita, De-Qi Yuan, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Photochem. Photobiol. A: Chem., 2005, 173, 375–383.
Enantiodifferentiating [4+ 4] photocyclodimerization of 2-anthracenecarboxylate catalyzed by 6A, 6X-diamino-6A, 6X-dideoxy-h-cyclodextrins: Manipulation of product chirality by electrostatic interaction, temperature and solvent in supramolecular photochirogenesisCheng Yang, Gaku Fukuhara, Asao Nakamura, Yumi Origane, Kahee Fujita, De-Qi Yuan, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Photochem. Photobiol. A: Chem., 2005, 173, 375–383.6A,6X-Dideoxy-6A,6X-diamino-γ-cyclodextrins (X = B, C, D and E) 5a–d were synthesized as chiral hosts for catalyzing the enantiodifferentiating [4 + 4] photocyclodimerization of 2-anthracenecarboxylic acid (ACA). The electrostatic interaction between 5a–d and ACA efficiently affected the preorganization of two ACA molecules within the γ-CD cavity, and improved the yields of head-to-head cyclodimers. By lowering the reaction temperature or solvent polarity, the electrostatic interaction was further enhanced. The anti-to-syn ratio of the head-to-head isomers gradually increased by changing the host from 5a to 5d (with increasing distance between the two amino groups on the CD rim), demonstrating a good structure–function relationship in this supramolecular photoreaction system. The chiral sense and enantiomeric excess of the photoproducts obtained are significantly affected, and even inverted, by solvent composition and reaction temperature. This temperature- and solvent-controlled chirality switching behavior is proven to originate from the contribution of non-zero differential entropy (ΔΔS‡) in the enantiodifferentiating process. This finding is the first example of a chirality inversion driven by entropy-related factors, such as solvent and temperature, in a non-sensitized asymmetric photoreaction. The sign of ΔΔS‡ was switched by the composition of solvent and exhibited an excellent compensatory relationship against the differential enthalpy (ΔΔH‡), revealing that the photoenantiodifferentiation is governed not only by enthalpy but also by entropy, and also that the enantiodifferentiation mechanism does not vary throughout the whole system, irrespective of the condition changes.
@article{yang2005enantiodifferentiating, title = {Enantiodifferentiating [4+ 4] photocyclodimerization of 2-anthracenecarboxylate catalyzed by 6A, 6X-diamino-6A, 6X-dideoxy-h-cyclodextrins: Manipulation of product chirality by electrostatic interaction, temperature and solvent in supramolecular photochirogenesis}, author = {Yang, Cheng and Fukuhara, Gaku and Nakamura, Asao and Origane, Yumi and Fujita, Kahee and Yuan, De-Qi and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {J. Photochem. Photobiol. A: Chem.}, volume = {173}, number = {3}, pages = {375--383}, year = {2005}, publisher = {Elsevier}, doi = {10.1016/j.jphotochem.2005.04.017}, url = {https://doi.org/10.1016/j.jphotochem.2005.04.017}, dimensions = {true}, tab = {paper}, } -
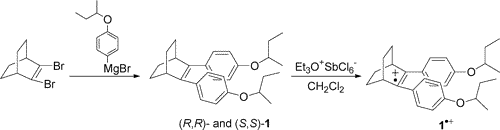 Chiral organic radical cation and dication. A reversible chiroptical redox switch based on stepwise transformation of optically active tetrakis (p-alkoxyphenyl) ethylenes to radical cations and dicationsTadashi Mori* and Yoshihisa Inoue*J. Phys. Chem. A, 2005, 109, 2728–2740.
Chiral organic radical cation and dication. A reversible chiroptical redox switch based on stepwise transformation of optically active tetrakis (p-alkoxyphenyl) ethylenes to radical cations and dicationsTadashi Mori* and Yoshihisa Inoue*J. Phys. Chem. A, 2005, 109, 2728–2740.Optically active tetrakis(p-alkoxyphenyl)ethylenes were found to function as reversible chiroptical switches upon redox transformations. Successive one-electron oxidations of chirally modified tetraarylethylene to the corresponding radical cation and then to the dication led to dramatic changes in the electronic absorption and circular dichroism (CD) spectra. The neutral species showed no color or CD in the visible region, while the radical ion was blue in color and exhibited a weak Cotton effect, with the dication green and giving an intense Cotton effect and a sign opposite that observed for the radical cation, at a longer wavelength. Molecular orbital calculations and X-ray crystallographic studies clearly indicate that the olefinic CC bond is significantly twisted in the dication to minimize the electrostatic and steric repulsions. By lowering the temperature of the dication, the twist around the double bond is more firmly fixed in either P or M chirality to give a stronger Cotton effect and a larger anisotropy (g) factor. Since the spectral changes are completely reversible and reproducible for multiple redox cycles, this chiral redox system can be used in novel redox-driven chiroptical applications, such as molecular switches and memory devices, in which the information is written/read chiroptically in the ternary mode, giving zero CD signal in the neutral form, positive CD for the radical cation, and negative CD for the dication at a given wavelength.
@article{mori2005chiral, title = {Chiral organic radical cation and dication. A reversible chiroptical redox switch based on stepwise transformation of optically active tetrakis (p-alkoxyphenyl) ethylenes to radical cations and dications}, author = {Mori, Tadashi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. A}, volume = {109}, number = {12}, pages = {2728--2740}, year = {2005}, publisher = {ACS Publications}, doi = {10.1021/jp044917m}, url = {https://doi.org/10.1021/jp044917m}, dimensions = {true}, tab = {paper}, } -
 Entropy-controlled supramolecular photochirogenesis: enantiodifferentiating Z–E photoisomerization of cyclooctene included and sensitized by permethylated 6-O-benzoyl-β-cyclodextrinGaku Fukuhara, Tadashi Mori, Takehiko Wada, and Yoshihisa InoueChem. Commun., 2005, 109, 4199–4201.
Entropy-controlled supramolecular photochirogenesis: enantiodifferentiating Z–E photoisomerization of cyclooctene included and sensitized by permethylated 6-O-benzoyl-β-cyclodextrinGaku Fukuhara, Tadashi Mori, Takehiko Wada, and Yoshihisa InoueChem. Commun., 2005, 109, 4199–4201.In contrast to the photosensitization with a non-methylated analogue, supramolecular photochirogenesis with a novel permethylated mono(6-O-benzoyl)-β-cyclodextrin exhibited a critical dependence of the product’s enantiomeric excess upon temperature, and possessed a large differential entropy of activation (−11 J K−1 mol−1) for which the flexible host skeleton is likely to be responsible.
@article{fukuhara2005entropy, title = {Entropy-controlled supramolecular photochirogenesis: enantiodifferentiating Z--E photoisomerization of cyclooctene included and sensitized by permethylated 6-O-benzoyl-β-cyclodextrin}, author = {Fukuhara, Gaku and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Commun.}, number = {33}, pages = {4199--4201}, year = {2005}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b504948b}, url = {https://doi.org/10.1039/b504948b}, dimensions = {true}, tab = {paper}, } - New Paradigm of Electrophilic Substitution MechanismTadashi Mori and Yoshihisa InoueKagaku, 2005, 60, 76–77.
@article{mori2005new, title = {New Paradigm of Electrophilic Substitution Mechanism}, author = {Mori, Tadashi and Inoue, Yoshihisa}, journal = {Kagaku}, volume = {60}, issue = {4}, pages = {76--77}, year = {2005} }
2004
-
 Pressure control of diastereodifferentiating [2+ 2] photocycloaddition of (E)-stilbene to chiral fumarate upon direct and charge-transfer excitationHideaki Saito, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2004, 60, 1652–1653.
Pressure control of diastereodifferentiating [2+ 2] photocycloaddition of (E)-stilbene to chiral fumarate upon direct and charge-transfer excitationHideaki Saito, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2004, 60, 1652–1653.The geometry (donor–acceptor distance) and association constant of the ground-state CT complex were considerably affected by applied pressure, thus enhancing the reaction rate and the adduct yield particularly upon CT excitation, but the diastereoselectivity did not show any substantial change at elevated pressures in both excitation modes.
@article{saito2004pressure, title = {Pressure control of diastereodifferentiating [2+ 2] photocycloaddition of (E)-stilbene to chiral fumarate upon direct and charge-transfer excitation}, author = {Saito, Hideaki and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Commun.}, number = {14}, pages = {1652--1653}, year = {2004}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b404555f}, url = {https://doi.org/10.1039/b404555f}, dimensions = {true}, tab = {paper}, } -
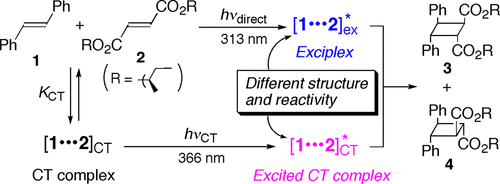 Diastereoselective [2+ 2] photocycloaddition of stilbene to chiral fumarate. Direct versus charge-transfer excitationHideaki Saito, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*J. Am. Chem. Soc., 2004, 126, 1900–1906.
Diastereoselective [2+ 2] photocycloaddition of stilbene to chiral fumarate. Direct versus charge-transfer excitationHideaki Saito, Tadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*J. Am. Chem. Soc., 2004, 126, 1900–1906.The selective excitation of the charge-transfer (CT) complex and the direct excitation of the substrate gave distinctly different product ratios and diastereomeric excesses (de’s), as well as their temperature dependencies, in [2 + 2] photocycloaddition of (E)-stilbene to bis((R)-1-methylpropyl) fumarate, clearly demonstrating that the excited CT complex and the conventional exciplex differ in structure and reactivity. This conclusion is supported by the contrasting fluorescence behavior exhibited by the relevant excited species, particularly at low temperatures.
@article{saito2004diastereoselective, title = {Diastereoselective [2+ 2] photocycloaddition of stilbene to chiral fumarate. Direct versus charge-transfer excitation}, author = {Saito, Hideaki and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {126}, number = {6}, pages = {1900--1906}, year = {2004}, publisher = {ACS Publications}, doi = {10.1021/ja0370140}, url = {https://doi.org/10.1021/ja0370140}, dimensions = {true}, tab = {paper}, } -
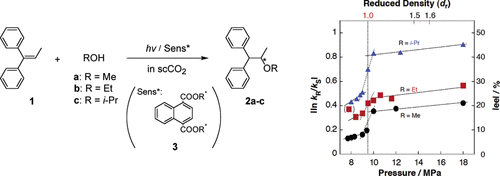 Enantiodifferentiating photoaddition of alcohols to 1, 1-diphenylpropene in supercritical carbon dioxide: Sudden jump of optical yield at the critical densityYasuhiro Nishiyama, Masayuki Kaneda, Ryota Saito, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Am. Chem. Soc., 2004, 126, 6568–6569.
Enantiodifferentiating photoaddition of alcohols to 1, 1-diphenylpropene in supercritical carbon dioxide: Sudden jump of optical yield at the critical densityYasuhiro Nishiyama, Masayuki Kaneda, Ryota Saito, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*J. Am. Chem. Soc., 2004, 126, 6568–6569.In the enantiodifferentiating photoaddition of ROH (R = Me, Et, i-Pr) to 1,1-diphenylpropene sensitized by fructosyl 1,4-naphthalenedicarboxylate in supercritical carbon dioxide, the enantiomeric excess of photoadduct increased with increasing bulkiness of the alcohol at all pressures used, with an accompanying sudden jump at the critical density, for which the enhanced clustering of alcohol, particularly in the subcritical pressure region, was revealed to be responsible from the fluorescence spectral examinations.
@article{nishiyama2004enantiodifferentiating, title = {Enantiodifferentiating photoaddition of alcohols to 1, 1-diphenylpropene in supercritical carbon dioxide: Sudden jump of optical yield at the critical density}, author = {Nishiyama, Yasuhiro and Kaneda, Masayuki and Saito, Ryota and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {126}, number = {21}, pages = {6568--6569}, year = {2004}, publisher = {ACS Publications}, doi = {10.1021/ja0494609}, url = {https://doi.org/10.1021/ja0494609}, dimensions = {true}, tab = {paper}, } -
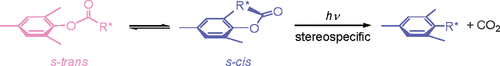 Mediation of conformationally controlled photodecarboxylations of chiral and cyclic aryl esters by substrate structure, temperature, pressure, and medium constraintsTadashi Mori*, Richard G Weiss, and Yoshihisa Inoue*J. Am. Chem. Soc., 2004, 126, 8961–8975.
Mediation of conformationally controlled photodecarboxylations of chiral and cyclic aryl esters by substrate structure, temperature, pressure, and medium constraintsTadashi Mori*, Richard G Weiss, and Yoshihisa Inoue*J. Am. Chem. Soc., 2004, 126, 8961–8975.An aryl alkanoate, 2,4,6-trimethylphenyl (S)-(+)-2-methylbutyrate, whose ester group has a chiral center alpha to the carbonyl carbon and in which photo-Fries rearrangements are blocked by methyl substituents, undergoes facile photodecarboxylation under a variety of conditions and with complete retention of configuration. In fact, the decarboxylation process has many of the attributes of a symmetry-allowed suprafacial [1,3]sigmatropic rearrangement. The process requires concerted extrusion of carbon dioxide in a spiro-lactonic transition state, which has been investigated using high level DFT and CIS calculations: thermally less stable s-cis conformers in the ground and excited singlet states play an important role in determining the competitive efficiency of the process. Conformational control has also been imposed by substrate structure, solvent interactions, temperature, and applying external pressure, as well as using constraining media such as cyclodextrins and polyethylene films. The results are correlated with steady-state and dynamic fluorescence measurements at various temperatures in order to investigate further degrees to which ground and excited singlet state conformations affect the different photoreactivity channels available to the aryl esters.
@article{mori2004mediation, title = {Mediation of conformationally controlled photodecarboxylations of chiral and cyclic aryl esters by substrate structure, temperature, pressure, and medium constraints}, author = {Mori, Tadashi and Weiss, Richard G and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {126}, number = {29}, pages = {8961--8975}, year = {2004}, publisher = {ACS Publications}, doi = {10.1021/ja049688w}, url = {https://doi.org/10.1021/ja049688w}, dimensions = {true}, tab = {paper}, } -
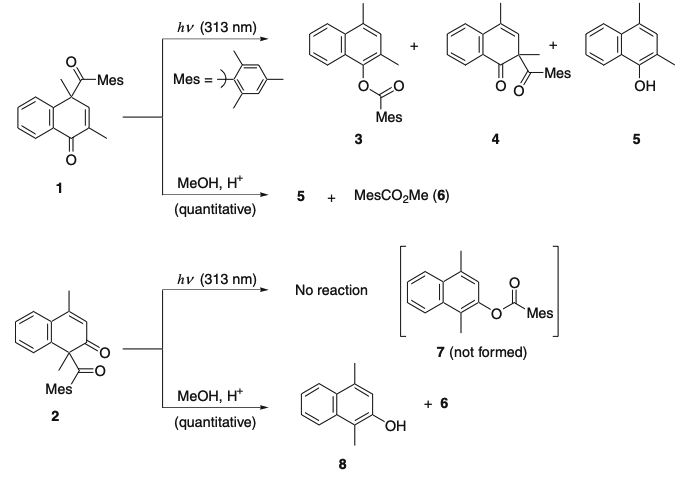 Remarkable differences in photo and thermal (acid-catalyzed) reactivities between ortho-and para-acylcyclohexadienones as essential factors determining the overall efficiency of the photo-Fries rearrangementTadashi Mori*, Makoto Takamoto, Hideaki Saito, Takahiro Furo, Takehiko Wada, and Yoshihisa Inoue*Chem. Lett., 2004, 33, 256–257.
Remarkable differences in photo and thermal (acid-catalyzed) reactivities between ortho-and para-acylcyclohexadienones as essential factors determining the overall efficiency of the photo-Fries rearrangementTadashi Mori*, Makoto Takamoto, Hideaki Saito, Takahiro Furo, Takehiko Wada, and Yoshihisa Inoue*Chem. Lett., 2004, 33, 256–257.Successful isolation of “ortho” and “para”-acylcyclohexadienones allowed us to comparatively study their ground- and excited-state behavior under a variety of conditions. In neutral solutions, the two isomeric cyclohexdienones showed completely different reactivities for photochemical and thermal reactions, while in acidic methanol both quantitatively afforded the corresponding transesterification product and naphthol. These studies help us understand the detailed photo-Fries rearrangement mechanism, which involves several crucial photochemical and thermal steps.
@article{mori2004remarkable, title = {Remarkable differences in photo and thermal (acid-catalyzed) reactivities between ortho-and para-acylcyclohexadienones as essential factors determining the overall efficiency of the photo-Fries rearrangement}, author = {Mori, Tadashi and Takamoto, Makoto and Saito, Hideaki and Furo, Takahiro and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Lett.}, volume = {33}, number = {3}, pages = {256--257}, year = {2004}, publisher = {Oxford University Press}, doi = {10.1246/cl.2004.256}, url = {https://doi.org/10.1246/cl.2004.256}, dimensions = {true}, tab = {paper}, } -
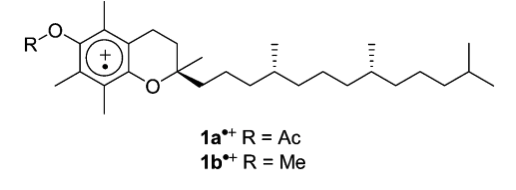 Chiroptical properties of organic radical cations. The electronic and vibrational circular dichroism spectra of α-tocopherol derivatives and sterically hindered chiral hydroquinone ethersTadashi Mori*, Hiroshi Izumi, and Yoshihisa Inoue*J. Phys. Chem. A, 2004, 108, 9540–9549.
Chiroptical properties of organic radical cations. The electronic and vibrational circular dichroism spectra of α-tocopherol derivatives and sterically hindered chiral hydroquinone ethersTadashi Mori*, Hiroshi Izumi, and Yoshihisa Inoue*J. Phys. Chem. A, 2004, 108, 9540–9549.Qualitatively and quantitatively reliable electronic and vibrational circular dichroism (ECD and VCD) spectra of chiral organic radical cations were obtained for the first time with α-tocopherol derivatives and sterically hindered chiral hydroquinone ethers. The isolation and spectral measurements of chiral radical cation salts were made possible by using nitrosonium or antimony derivatives as electron-transfer oxidants, which can cleanly oxidize the substrate donors without giving any byproducts in the sample solution. Such reliable ECD spectra enabled us to fully examine the chiroptical properties of organic radical cations and also compare them with those of the corresponding neutral compounds. The observed VCD spectra of neutral and radical cationic species of chiral hydroquinone ether were nicely simulated by density functional theory (DFT) calculations, from which the relative contribution of each radical cation conformer in solution was evaluated. Thus, the combined synthetic, spectroscopic, and theoretical protocol, composed of chiral modification, clean oxidation to form stable radical cations, ECD/VCD spectral analyses, and DFT calculations, was demonstrated to be a powerful, indispensable tool for elucidating a comprehensive picture of radical cationic species in solution.
@article{mori2004chiroptical, title = {Chiroptical properties of organic radical cations. The electronic and vibrational circular dichroism spectra of $\alpha$-tocopherol derivatives and sterically hindered chiral hydroquinone ethers}, author = {Mori, Tadashi and Izumi, Hiroshi and Inoue, Yoshihisa}, journal = {J. Phys. Chem. A}, volume = {108}, number = {44}, pages = {9540--9549}, year = {2004}, publisher = {ACS Publications}, doi = {10.1021/jp0463520}, url = {https://doi.org/10.1021/jp0463520}, dimensions = {true}, tab = {paper}, } -
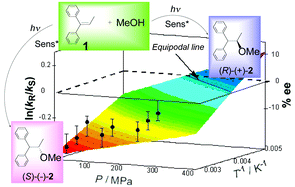 Pressure control of enantiodifferentiating polar addition of 1, 1-diphenylpropene sensitized by chiral naphthalenecarboxylatesMasayuki Kaneda, Yasuhiro Nishiyama, Sadayuki Asaoka, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Org. Biomol. Chem., 2004, 2, 1295–1303.
Pressure control of enantiodifferentiating polar addition of 1, 1-diphenylpropene sensitized by chiral naphthalenecarboxylatesMasayuki Kaneda, Yasuhiro Nishiyama, Sadayuki Asaoka, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Org. Biomol. Chem., 2004, 2, 1295–1303.Effects of pressure on the enantiodifferentiating methanol addition to 1,1-diphenylpropene (1) sensitized by chiral naphthalenedicarboxylates (3 and 4) were investigated over 0.1–400 MPa. The logarithm of enantiomeric excess (ee) of photoadduct, i.e. 1,1-diphenyl-2-methoxypropane (2), was a linear function of both pressure (P) and temperature (T); further, the product chirality was switched by P in some cases. From the slope of P − ln(kR/kS) plot, the differential activation volume (ΔΔV‡) was determined for the first time for bimolecular asymmetric photoreactions. The ΔΔV‡ values obtained are mostly larger than those obtained for relevant unimolecular photoreactions, and are a critical function of the nature of the chiral auxiliary and solvent, indicating conformation changes of the intervening diastereomeric exciplex or transition state in different solvents. Indeed, fluorescence spectral examinations of the sensitizer and exciplex under high pressure revealed the existence of exciplexes of variable energy and structure, which may rationalize the different ΔΔV‡ and product ee obtained. A three-dimensional diagram, correlating the ee with P and T, was constructed from the pressure dependence data at different T, from which we may propose an idea of the multidimensional control of asymmetric reaction by the combined use of the entropy-related enviromental factors.
@article{kaneda2004pressure, title = {Pressure control of enantiodifferentiating polar addition of 1, 1-diphenylpropene sensitized by chiral naphthalenecarboxylates}, author = {Kaneda, Masayuki and Nishiyama, Yasuhiro and Asaoka, Sadayuki and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Org. Biomol. Chem.}, volume = {2}, number = {9}, pages = {1295--1303}, year = {2004}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b401108b}, url = {https://doi.org/10.1039/b401108b}, dimensions = {true}, tab = {paper}, } - Anion Radical (Cation Radical) π-Mer and Dimer Dianion (Dication)Tadashi MoriKokagaku, 2004, 35, 51–53.
@article{mori2004anion, title = {Anion Radical (Cation Radical) π-Mer and Dimer Dianion (Dication)}, author = {Mori, Tadashi}, journal = {Kokagaku}, volume = {35}, issue = {1}, pages = {51--53}, year = {2004} }
2003
-
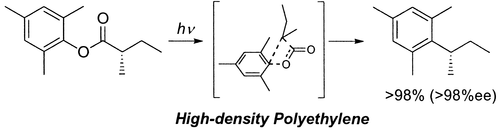 Enhanced photodecarboxylation of an aryl ester in polyethylene filmsTadashi Mori*, Yoshihisa Inoue, and Richard G Weiss*Org. Lett., 2003, 5, 4661–4664.
Enhanced photodecarboxylation of an aryl ester in polyethylene filmsTadashi Mori*, Yoshihisa Inoue, and Richard G Weiss*Org. Lett., 2003, 5, 4661–4664.Photodecarboxylation is the exclusive photoreaction of 2,4,6-trimethylphenyl (S)-2-methylbutanoate in unstretched high-density polyethylene films. The sole product, (S)-1-(2-methylpropyl)-2,4,6-trimethylbenzene, is formed with complete retention of stereochemistry. In other polyethylene films, organic solvents, and β-cyclodextrin cavities, cage-escaped products derived from Fries-type bond scission are obtained as well. The results indicate the importance of the media in controlling the conformations of aryl esters and, thereby, their photoreactions.
@article{mori2003enhanced, title = {Enhanced photodecarboxylation of an aryl ester in polyethylene films}, author = {Mori, Tadashi and Inoue, Yoshihisa and Weiss, Richard G}, journal = {Org. Lett.}, volume = {5}, number = {24}, pages = {4661--4664}, year = {2003}, publisher = {ACS Publications}, doi = {10.1021/ol035852t}, url = {https://doi.org/10.1021/ol035852t}, dimensions = {true}, tab = {paper}, } -
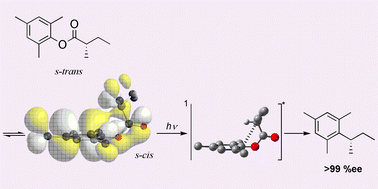 Complete memory of chirality upon photodecarboxylation of mesityl alkanoate to mesitylalkane: theoretical and experimental evidence for cheletropic decarboxylation via a spiro-lactonic transition stateTadashi Mori*, Hideaki Saito, and Yoshihisa Inoue*Chem. Commun., 2003, 5, 2302–2303.
Complete memory of chirality upon photodecarboxylation of mesityl alkanoate to mesitylalkane: theoretical and experimental evidence for cheletropic decarboxylation via a spiro-lactonic transition stateTadashi Mori*, Hideaki Saito, and Yoshihisa Inoue*Chem. Commun., 2003, 5, 2302–2303.The photodecarboxylation of chiral mesityl alkanoate to mesitylalkane has been studied experimentally/theoretically, and it has been found that the photodecarboxylation proceeds to give the product in >99% enantiomeric excesses under a variety of conditions, indicating no involvement of any radical intermediates, but that the reaction proceeds through the concerted cheletropic extrusion of CO2 from the energetically less-favored s-cis conformation.
@article{mori2003complete, title = {Complete memory of chirality upon photodecarboxylation of mesityl alkanoate to mesitylalkane: theoretical and experimental evidence for cheletropic decarboxylation via a spiro-lactonic transition state}, author = {Mori, Tadashi and Saito, Hideaki and Inoue, Yoshihisa}, journal = {Chem. Commun.}, number = {18}, pages = {2302--2303}, year = {2003}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b305267b}, url = {https://doi.org/10.1039/b305267b}, dimensions = {true}, tab = {paper}, } -
 Bovine serum albumin-mediated enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylateTakehiko Wada, Masaki Nishijima, Tai Fujisawa, Norimitsu Sugahara, Tadashi Mori, Asao Nakamura, and Yoshihisa Inoue*J. Am. Chem. Soc., 2003, 125, 7492–7493.
Bovine serum albumin-mediated enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylateTakehiko Wada, Masaki Nishijima, Tai Fujisawa, Norimitsu Sugahara, Tadashi Mori, Asao Nakamura, and Yoshihisa Inoue*J. Am. Chem. Soc., 2003, 125, 7492–7493.Enantiodifferentiating photocyclodimerization of 2-anthracenecarboxyalate (AC) was performed at 25 °C in aqueous buffer solution (pH 7) in the presence of bovine-serum albumin (BSA) to afford four [4 + 4] cyclodimers, i.e., anti- and syn-head-to-tail (HT) (1 and 2) and anti- and syn-head-to-head (HH) dimers (3 and 4), of which only 2 and 3 are chiral. We found that (1) BSA possesses four sets of binding sites for AC of different affinities, stoichiometries, and chiral environment for photoreaction, which bind 1, 3, 2, and 3 AC molecules with binding constants of 5.3 × 107, 1.3 × 105, 1.4 × 104, and 3.0 × 103 M-1, respectively, (2) the regioselectivity of photodimerization is switched from HT to HH by adding BSA (the HH/HT ratio varies from 0.28 to 4.3), (3) BSA-mediated photodimerization of AC affords optically active products 2 and 3 of up to 29% and 41% ee, respectively. It is emphasized that the selective excitation of bound substrate, utilizing the spectral shift upon complexation with BSA, is not a prerequisite for efficient photochirogenesis using biomolecules.
@article{wada2003bovine, title = {Bovine serum albumin-mediated enantiodifferentiating photocyclodimerization of 2-anthracenecarboxylate}, author = {Wada, Takehiko and Nishijima, Masaki and Fujisawa, Tai and Sugahara, Norimitsu and Mori, Tadashi and Nakamura, Asao and Inoue, Yoshihisa}, journal = {J. Am. Chem. Soc.}, volume = {125}, number = {25}, pages = {7492--7493}, year = {2003}, publisher = {ACS Publications}, doi = {10.1021/ja034641g}, url = {https://doi.org/10.1021/ja034641g}, dimensions = {true}, tab = {paper}, } -
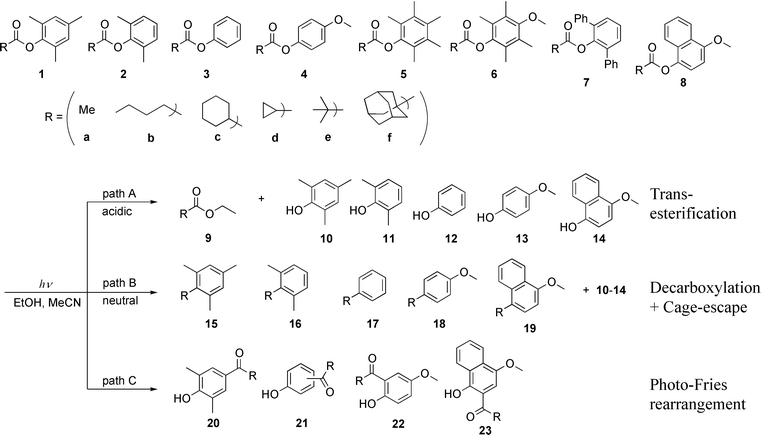 Acid-controlled photoreactions of aryl alkanoates: competition of transesterification, decarboxylation, Fries-rearrangement and/or transpositionTadashi Mori*, Makoto Takamoto, Takehiko Wada, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2003, 2, 1187–1199.
Acid-controlled photoreactions of aryl alkanoates: competition of transesterification, decarboxylation, Fries-rearrangement and/or transpositionTadashi Mori*, Makoto Takamoto, Takehiko Wada, and Yoshihisa Inoue*Photochem. Photobiol. Sci., 2003, 2, 1187–1199.2,4,6-Trimethylphenyl (mesityl) cyclohexanecarboxylate 1c and related mesityl esters 1a,b and d–f were photodecarboxylated upon irradiation at 254 nm in neutral solvents to give alkylmesitylenes 15 in good yields. In contrast, in the presence of a catalytic amount of acid and a sufficient amount of alcohol, the same compounds underwent facile phototransesterification to afford the corresponding ester 9 and phenol 10 in almost quantitative yields. Photolyses of less-substituted aryl alkanoates such as 2,6-dimethylphenyl (xylenyl), phenyl, 4-methoxyphenyl (4-anisyl) and 4-methoxynaphthyl alkanoates 2–4 and 8 were also investigated to elucidate the reaction mechanism of acid-catalyzed phototransesterification. Irradiation of these esters afforded the corresponding photo-Fries rearrangement products 20–23 both in neutral and acidic acetonitrile with significant acceleration of the processes in the presence of acid. It was elucidated that the photolysis of the esters affords the geminate radical pair 28, which in turn recombines to cyclohexadienone intermediates 29 and 30. Added acid accelerates not only the nucleophilic attack of alcohol to 29 and 30 but also other processes. Prolonged irradiation of the esters in neutral solution led to skeletal rearrangements of the initial product, affording isomeric alkylbenzenes. The phototransposition of cyclohexylmesitylene 15cvia benzvalene intermediates was, on the contrary, retarded under acidic conditions. These acid-controlled competitive photoreactions are representative examples, in which a catalytic amount of acid can alter the fate of reactive intermediates on both ground-state and excited-state surfaces.
@article{mori2003acid, title = {Acid-controlled photoreactions of aryl alkanoates: competition of transesterification, decarboxylation, Fries-rearrangement and/or transposition}, author = {Mori, Tadashi and Takamoto, Makoto and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Photochem. Photobiol. Sci.}, volume = {2}, number = {11}, pages = {1187--1199}, year = {2003}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b305898k}, url = {https://doi.org/10.1039/b305898k}, dimensions = {true}, tab = {paper}, } -
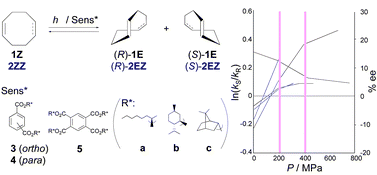 Pressure control of enantiodifferentiating photoisomerization of cyclooctenes sensitized by chiral benzenepolycarboxylates. The origin of discontinuous pressure dependence of the optical yieldMasayuki Kaneda, Asao Nakamura, Sadayuki Asaoka, Haruhiko Ikeda, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Org. Biomol. Chem., 2003, 1, 4435–4440.
Pressure control of enantiodifferentiating photoisomerization of cyclooctenes sensitized by chiral benzenepolycarboxylates. The origin of discontinuous pressure dependence of the optical yieldMasayuki Kaneda, Asao Nakamura, Sadayuki Asaoka, Haruhiko Ikeda, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Org. Biomol. Chem., 2003, 1, 4435–4440.Pressure effects on enantiodifferentiating geometrical photoisomerizations of (Z)-cyclooctene and (Z,Z)-cycloocta-1,5-diene sensitized by chiral benzene-1,2,4,5-tetracarboxylate were investigated over a pressure range of 0.1–750 MPa. Enantiomeric excesses (ee’s) of the (E)- and (E,Z)-isomers obtained displayed discontinuous pressure dependencies, affording distinctly different differential activation volumes (ΔΔV‡) for each range, indicating alteration of the enantiodifferentiation mechanism. The switching of ΔΔV‡ occurred at essentially the same pressures of 200 and 400 MPa, which are shared by all the chiral sensitizers, irrespective of the chiral auxiliary employed. Circular dichroism spectral examinations at pressures of up to 400 MPa also revealed that the chiral sensitizers undergo discontinuous conformational changes at 200 MPa, which most likely lead to switching of the enantiodifferentiating sensitization mechanism in the exciplex intermediate.
@article{kaneda2003pressure, title = {Pressure control of enantiodifferentiating photoisomerization of cyclooctenes sensitized by chiral benzenepolycarboxylates. The origin of discontinuous pressure dependence of the optical yield}, author = {Kaneda, Masayuki and Nakamura, Asao and Asaoka, Sadayuki and Ikeda, Haruhiko and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Org. Biomol. Chem.}, volume = {1}, number = {24}, pages = {4435--4440}, year = {2003}, publisher = {Royal Society of Chemistry}, doi = {10.1039/b310491e}, url = {https://doi.org/10.1039/b310491e}, dimensions = {true}, tab = {paper}, }
2002
-
 Steric hindrance as a mechanistic probe for olefin reactivity: Variability of the hydrogenic canopy over the isomeric adamantylideneadamantane/sesquihomoadamantene pair (a combined experimental and theoretical study)Rajendra Rathore, Sergey V Lindeman, C-J Zhu, Tadashi Mori, P v R Schleyer, and Jay K Kochi*J. Org. Chem., 2002, 67, 5106–5116.
Steric hindrance as a mechanistic probe for olefin reactivity: Variability of the hydrogenic canopy over the isomeric adamantylideneadamantane/sesquihomoadamantene pair (a combined experimental and theoretical study)Rajendra Rathore, Sergey V Lindeman, C-J Zhu, Tadashi Mori, P v R Schleyer, and Jay K Kochi*J. Org. Chem., 2002, 67, 5106–5116.Access to each CC face of adamantylideneadamantane (AA) and sesquihomoadamantene (SA) is hindered by the hydrogenic canopy consisting of four β-hydrogens; otherwise, these olefins have quite normal environments. X-ray crystallography and density functional (DFT) calculations show a 0.5 Å larger annular opening in the protective cover of AA than that in SA. This contributes to the remarkable differences in reactivity toward various reagents, not only by limiting access to the olefin site in SA but also by inhibiting reactions which force these hydrogens closer together. Thus, AA is subject to typical olefin-addition reactions with bromine, sulfuryl chloride, m-chloroperbenzoic acid, dioxygen, and so forth, albeit sometimes at attenuated rates. On the other hand, SA is singularly unreactive under identical reaction conditions, except for the notable exceptions that include Brønsted (protonic) acids, a nitrosonium cation, and dichlorine. The exceptions are characterized as three sterically limited (electrophilic) reagents whose unique reactivity patterns are shown to be strongly influenced by steric access to the CC center. As such, the different degrees of steric encumbrance in the isomeric donors AA and SA shed considerable light on the diverse nature of olefinic reactions. In particular, they evoke mechanistic features in electrophilic addition versus electron transfer, which are otherwise not readily discernible with other less hindered olefinic donors. Transient structures of the olefinic-reaction intermediates such as the protonated carbocations AA−H+ and SA−H+ as well as the cation radicals AA•+ and SA•+ are probed by the combination of X-ray crystallographic analyses and density functional theoretical computations.
@article{rathore2002steric, title = {Steric hindrance as a mechanistic probe for olefin reactivity: Variability of the hydrogenic canopy over the isomeric adamantylideneadamantane/sesquihomoadamantene pair (a combined experimental and theoretical study)}, author = {Rathore, Rajendra and Lindeman, Sergey V and Zhu, C-J and Mori, Tadashi and Schleyer, P v R and Kochi, Jay K}, journal = {J. Org. Chem.}, volume = {67}, number = {15}, pages = {5106--5116}, year = {2002}, publisher = {ACS Publications}, doi = {10.1021/jo0200724}, url = {https://doi.org/10.1021/jo0200724}, dimensions = {true}, tab = {paper}, } -
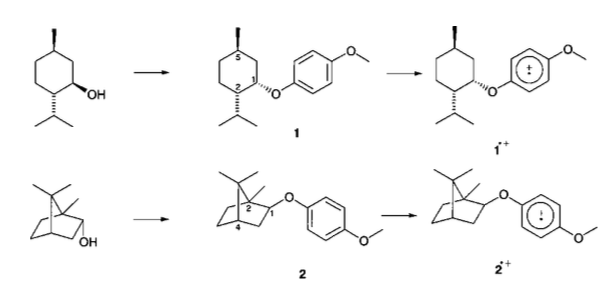 The first circular dichroism observation for organic radical cations: Chiroptical properties of neomenthyloxy-and isobornyloxyanisole radical cationsTadashi Mori, Junya Shinkuma, Masafumi Sato, Hideaki Saito, Takehiko Wada, and Yoshihisa Inoue*Enantiomer, 2002, 7, 115–118.
The first circular dichroism observation for organic radical cations: Chiroptical properties of neomenthyloxy-and isobornyloxyanisole radical cationsTadashi Mori, Junya Shinkuma, Masafumi Sato, Hideaki Saito, Takehiko Wada, and Yoshihisa Inoue*Enantiomer, 2002, 7, 115–118.The first unequivocal circular dichroism spectra were obtained for organic radical cations, which were prepared from p-(1S,2S,5R) -neomenthyloxyanisole (1) and p-(1S,2S,4S) -isobornyloxyanisole (2) by single electron oxidation with triethyloxonium hexachloroantimonate in dichloromethane. The chiroptical properties of the radical cations 1·+ and 2·+ were discussed in comparison with those of neutral species 1·+ and 2.
@article{mori2002first, title = {The first circular dichroism observation for organic radical cations: Chiroptical properties of neomenthyloxy-and isobornyloxyanisole radical cations}, author = {Mori, Tadashi and Shinkuma, Junya and Sato, Masafumi and Saito, Hideaki and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Enantiomer}, volume = {7}, number = {2-3}, pages = {115--118}, year = {2002}, publisher = {Taylor Francis}, doi = {10.1080/10242430212199}, url = {https://doi.org/10.1080/10242430212199}, dimensions = {true}, tab = {paper}, } -
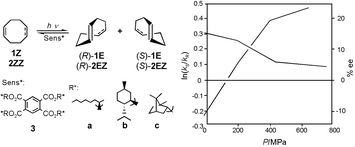 Discontinuous pressure effect upon enantiodifferentiating photosensitized isomerization of cycloocteneMasayuki Kaneda, Sadayuki Asaoka, Haruhiko Ikeda, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2002, 7, 1272–1273.
Discontinuous pressure effect upon enantiodifferentiating photosensitized isomerization of cycloocteneMasayuki Kaneda, Sadayuki Asaoka, Haruhiko Ikeda, Tadashi Mori, Takehiko Wada, and Yoshihisa Inoue*Chem. Commun., 2002, 7, 1272–1273.A hydrostatic pressure of up to 750 MPa induced discontinuous changes in the enantiomeric excess of the (E)-isomer obtained in the enantiodifferentiating photoisomerization of (Z)-cyclooctene and (Z,Z)-cycloocta-1,5-diene, sensitized by chiral benzene-1,2,4,5-tetracarboxylates; indicating a switching of the enantiodifferentiation mechanism, which is attributable to dramatic conformational changes of chiral alkoxycarbonyl auxiliaries at a specific pressure.
@article{kaneda2002discontinuous, title = {Discontinuous pressure effect upon enantiodifferentiating photosensitized isomerization of cyclooctene}, author = {Kaneda, Masayuki and Asaoka, Sadayuki and Ikeda, Haruhiko and Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Chem. Commun.}, number = {12}, pages = {1272--1273}, year = {2002}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/b202699f}, url = {https://doi.org/10.1039/b202699f}, dimensions = {true}, tab = {paper}, } -
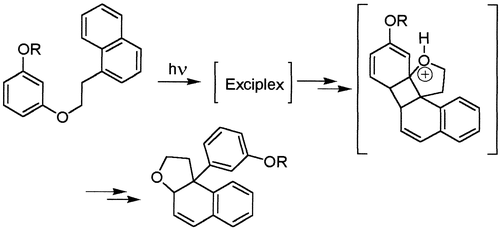 Novel [2+ 2] photocycloaddition-induced rearrangement of bichromophoric naphthalene-tethered resorcinol ethersNorbert Hoffmann*, Jean-Pierre Pete, Yoshihisa Inoue*, and Tadashi Mori*J. Org. Chem., 2002, 67, 2315–2322.
Novel [2+ 2] photocycloaddition-induced rearrangement of bichromophoric naphthalene-tethered resorcinol ethersNorbert Hoffmann*, Jean-Pierre Pete, Yoshihisa Inoue*, and Tadashi Mori*J. Org. Chem., 2002, 67, 2315–2322.The first examples of sequential photocycloaddition−rearrangement reactions of naphthalene-tethered resorcinol ethers are described. Bichromophoric aromatic compounds with naphthalene and resorcinol ether moieties were irradiated in the presence/absence of a small amount of acid to give the corresponding cycloaddition−rearrangement products. From the determination of quantum yields, steady-state fluorescence spectral studies, and fluorescence lifetime measurements, the mechanism of this novel photoinduced multistep reaction was elucidated to involve the initial intramolecular exciplex formation, followed by the intramolecular [2 + 2] photocycloaddition between the two aromatic rings and the subsequent acid-catalyzed skeletal rearrangement of the resulting cyclobutane derivative leading to the final products.
@article{hoffmann2002novel, title = {Novel [2+ 2] photocycloaddition-induced rearrangement of bichromophoric naphthalene-tethered resorcinol ethers}, author = {Hoffmann, Norbert and Pete, Jean-Pierre and Inoue, Yoshihisa and Mori, Tadashi}, journal = {J. Org. Chem.}, volume = {67}, number = {7}, pages = {2315--2322}, year = {2002}, publisher = {ACS Publications}, doi = {10.1021/jo011143m}, url = {https://doi.org/10.1021/jo011143m}, dimensions = {true}, tab = {paper}, }
2001
-
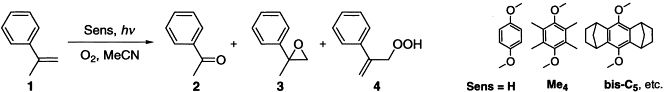 Photoinduced electron transfer oxidation of α-methylstyrene with molecular oxygen sensitized by dimethoxybenzenes: a non-singlet-oxygen mechanismTadashi Mori*, Makoto Takamoto, Yoshimasa Tate, Junya Shinkuma, Takehiko Wada, and Yoshihisa Inoue*Tetrahedron Lett., 2001, 42, 2505–2508.
Photoinduced electron transfer oxidation of α-methylstyrene with molecular oxygen sensitized by dimethoxybenzenes: a non-singlet-oxygen mechanismTadashi Mori*, Makoto Takamoto, Yoshimasa Tate, Junya Shinkuma, Takehiko Wada, and Yoshihisa Inoue*Tetrahedron Lett., 2001, 42, 2505–2508.α-Methylstyrene (1) was photooxidized with oxygen in the presence of a series of alkylated dimethoxybenzenes as a sensitizer in acetonitrile, affording the cleaved ketone (2), epoxide (3) as well as a small amount of the ene product (4) in ca. 1:1:0.04 ratio. A non-singlet-oxygen mechanism is proposed, in which an excited sensitizer is quenched by oxygen to produce a sensitizer radical cation and a superoxide ion (O2radical dot−), the former of which oxidizes 1, while O2radical dot− reacts with the resulting 1radical dot+ to give the major oxidation products.
@article{mori2001photoinduced, title = {Photoinduced electron transfer oxidation of α-methylstyrene with molecular oxygen sensitized by dimethoxybenzenes: a non-singlet-oxygen mechanism}, author = {Mori, Tadashi and Takamoto, Makoto and Tate, Yoshimasa and Shinkuma, Junya and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Tetrahedron Lett.}, volume = {42}, number = {13}, pages = {2505--2508}, year = {2001}, publisher = {Elsevier}, doi = {10.1016/S0040-4039(01)00213-1}, url = {https://doi.org/10.1016/S0040-4039(01)00213-1}, dimensions = {true}, tab = {paper}, } -
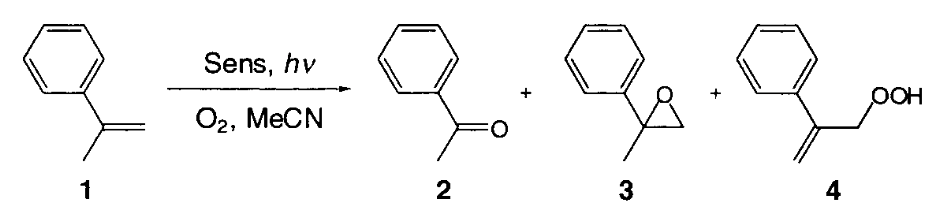 Photoinduced Electron-Transfer Oxidation of Olefins with Molecular Oxygen Sensitized by Tetrasubstituted Dimethoxybenzenes: A Non-Singlet-Oxygen MechanismTadashi Mori*, Makoto Takamoto, Takehiko Wada, and Yoshihisa Inoue*Helv. Chim. Acta, 2001, 84, 2693–2707.
Photoinduced Electron-Transfer Oxidation of Olefins with Molecular Oxygen Sensitized by Tetrasubstituted Dimethoxybenzenes: A Non-Singlet-Oxygen MechanismTadashi Mori*, Makoto Takamoto, Takehiko Wada, and Yoshihisa Inoue*Helv. Chim. Acta, 2001, 84, 2693–2707.α-Methylstyrene (1) was photo-oxidized in the presence of a series of alkylated dimethoxybenzenes as sensitizers in an oxygen-saturated MeCN solution to afford the cleaved ketone 2, epoxide 3, as well as a small amount of the ene product 4 in ca. 1: 1:0.04 ratio. The relative rate of conversion was well-correlated with the fluorescence quantum yield of sensitizers. Thus, a non-singlet-oxygen mechanism is proposed, in which an excited sensitizer is quenched by (ground-state) molecular oxygen to produce a sensitizer radical cation and a superoxide ion (O), the former of which oxidizes the substrate, while the latter reacts with the resulting olefin radical cation (1+.) to give the major oxidation products. Photodurability of such electron-donating sensitizers is dramatically improved by substituting four aromatic H-atoms in 1,4-dimethoxybenzene with Me or fused alkyl groups, which provides us with an environmentally friendly, clean method of photochemical functionalization with molecular oxygen, alternative to the ene reaction via singlet oxygenation.
@article{mori2001photoinduced2, title = {Photoinduced Electron-Transfer Oxidation of Olefins with Molecular Oxygen Sensitized by Tetrasubstituted Dimethoxybenzenes: A Non-Singlet-Oxygen Mechanism}, author = {Mori, Tadashi and Takamoto, Makoto and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Helv. Chim. Acta}, volume = {84}, number = {9}, pages = {2693--2707}, year = {2001}, publisher = {Wiley Online Library}, doi = {10.1002/1522-2675(20010919)84:9<2693::AID-HLCA2693>3.0.CO;2-3}, url = {https://doi.org/10.1002/1522-2675(20010919)84:9<2693::AID-HLCA2693>3.0.CO;2-3}, dimensions = {true}, tab = {paper}, }
2000
-
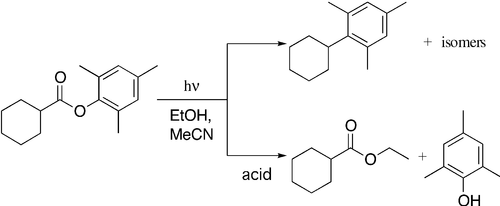 Perfect switching of photoreactivity by acid: Photochemical decarboxylation versus transesterification of mesityl cyclohexanecarboxylateTadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*Org. Lett., 2000, 2, 3401–3404.
Perfect switching of photoreactivity by acid: Photochemical decarboxylation versus transesterification of mesityl cyclohexanecarboxylateTadashi Mori*, Takehiko Wada, and Yoshihisa Inoue*Org. Lett., 2000, 2, 3401–3404.Mesityl cyclohexanecarboxylate has been photodecarboxylated upon excitation at 254 nm to form cyclohexylmesitylene in good yield in neutral acetonitrile solutions. In the presence of a catalytic amount of the acid and ethanol as a nucleophile, however, the same compound undergoes facile transesterification upon irradiation, affording its ethyl ester in almost quantitative yield.
@article{mori2000perfect, title = {Perfect switching of photoreactivity by acid: Photochemical decarboxylation versus transesterification of mesityl cyclohexanecarboxylate}, author = {Mori, Tadashi and Wada, Takehiko and Inoue, Yoshihisa}, journal = {Org. Lett.}, volume = {2}, number = {21}, pages = {3401--3404}, year = {2000}, publisher = {ACS Publications}, doi = {10.1021/ol0065266}, url = {https://doi.org/10.1021/ol0065266}, dimensions = {true}, tab = {paper}, }
1998
-
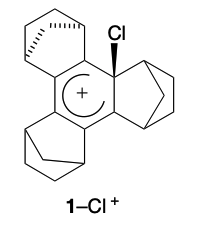 X-Ray structure of bridged 2, 2′-bi (adamant-2-ylidene) chloronium cation and comparison of its reactivity with a singly-bonded chloroarenium cationTadashi Mori, Rajendra Rathore, S V Lindeman, and J K Kochi*Chem. Commun., 1998, 2, 927–928.
X-Ray structure of bridged 2, 2′-bi (adamant-2-ylidene) chloronium cation and comparison of its reactivity with a singly-bonded chloroarenium cationTadashi Mori, Rajendra Rathore, S V Lindeman, and J K Kochi*Chem. Commun., 1998, 2, 927–928.The unsymmetrically bridged 2,2′-bi(adamant-2-ylidene) chloronium hexachloroantimonate shows distinctive electrophilic (transfer) chlorination reactivity in comparison with the singly-bonded chloroarenium cation.
@article{mori1998x, title = {X-Ray structure of bridged 2, 2′-bi (adamant-2-ylidene) chloronium cation and comparison of its reactivity with a singly-bonded chloroarenium cation}, author = {Mori, Tadashi and Rathore, Rajendra and Lindeman, S V and Kochi, J K}, journal = {Chem. Commun.}, number = {8}, pages = {927--928}, year = {1998}, publisher = {Royal Society of Chemistry}, doi = {10.1039/a709063c}, url = {https://doi.org/10.1039/a709063c}, dimensions = {true}, tab = {paper}, } -
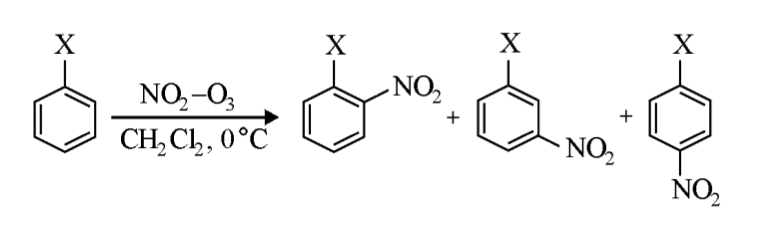 Kyodai-Nitration. An Alternative Electrophilic Route to Aromatic Nitro Compounds Based on Nitrogen TrioxideNobuaki Nonoyama, Tadashi Mori, and Hitomi Suzuki*Russ. J. Org. Chem., 1998, 34, 1521–1531.
Kyodai-Nitration. An Alternative Electrophilic Route to Aromatic Nitro Compounds Based on Nitrogen TrioxideNobuaki Nonoyama, Tadashi Mori, and Hitomi Suzuki*Russ. J. Org. Chem., 1998, 34, 1521–1531.@article{nonoyama1998kyodai, title = {Kyodai-Nitration. An Alternative Electrophilic Route to Aromatic Nitro Compounds Based on Nitrogen Trioxide}, author = {Nonoyama, Nobuaki and Mori, Tadashi and Suzuki, Hitomi}, journal = {Russ. J. Org. Chem.}, volume = {34}, issue = {11}, pages = {1521--1531}, year = {1998}, publisher = {Kluwer Academic Plenum Publisher}, doi = {10.1002/chin.199917310}, url = {https://doi.org/10.1002/chin.199917310}, dimensions = {true}, tab = {review}, }
1997
-
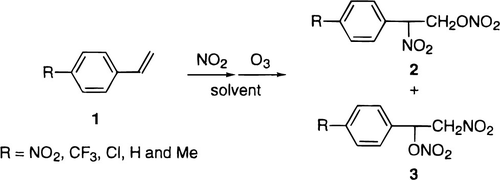 Side-chain nitration of styrene and para-substituted derivatives with a combination of nitrogen dioxide and ozoneHitomi Suzuki* and Tadashi MoriJ. Org. Chem., 1997, 62, 6498–6502.
Side-chain nitration of styrene and para-substituted derivatives with a combination of nitrogen dioxide and ozoneHitomi Suzuki* and Tadashi MoriJ. Org. Chem., 1997, 62, 6498–6502.The side-chain nitration of styrene and four para-substituted derivatives 1 with a combination of nitrogen dioxide and ozone has been investigated as part of our continued effort to define the scope of applicability of this new nitration methodology (kyodai nitration). Styrene and derivatives underwent a smooth addition reaction across the olefinic bond in dichloromethane at −20 °C, affording the corresponding isomeric nitro-nitrato adducts 2 and 3 in excellent combined yield. 1H-NMR data were collected for all possible nitro-nitrato adducts of styrene and derivatives. The mechanism of the formation of these addition products is discussed in terms of the reversible addition of nitrogen dioxide to form unstable nitroso-nitrates 9 and 10 followed by rapid oxidation of these to the corresponding nitro-nitrates 2 and 3.
@article{suzuki1997side, title = {Side-chain nitration of styrene and para-substituted derivatives with a combination of nitrogen dioxide and ozone}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {J. Org. Chem.}, volume = {62}, number = {19}, pages = {6498--6502}, year = {1997}, publisher = {ACS Publications}, doi = {10.1021/jo9705024}, url = {https://doi.org/10.1021/jo9705024}, dimensions = {true}, tab = {paper}, } -
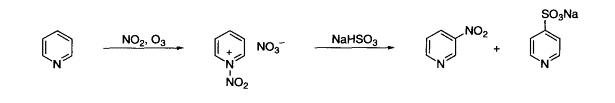 C-Nitration of pyridine by the kyodai-nitration modified by the Bakke procedure. A simple route to 3-nitropyridine and mechanistic aspect of its formationHitomi Suzuki*, Masao Iwaya, and Tadashi MoriTetrahedron Lett., 1997, 38, 5647–5650.
C-Nitration of pyridine by the kyodai-nitration modified by the Bakke procedure. A simple route to 3-nitropyridine and mechanistic aspect of its formationHitomi Suzuki*, Masao Iwaya, and Tadashi MoriTetrahedron Lett., 1997, 38, 5647–5650.N-Nitropyridinium nitrate was generated in situ from pyridine, nitrogen dioxide and ozone in an inert organic solvent and subsequently treated with aqueous sodium hydrogen sulfite to afford 3-nitropyridine in good yield, together with sodium pyridine-4-sulfonate as a water-soluble by-product.
@article{suzuki1997c, title = {C-Nitration of pyridine by the kyodai-nitration modified by the Bakke procedure. A simple route to 3-nitropyridine and mechanistic aspect of its formation}, author = {Suzuki, Hitomi and Iwaya, Masao and Mori, Tadashi}, journal = {Tetrahedron Lett.}, volume = {38}, number = {32}, pages = {5647--5650}, year = {1997}, publisher = {Elsevier}, doi = {10.1016/S0040-4039(97)01229-X}, url = {https://doi.org/10.1016/S0040-4039(97)01229-X}, dimensions = {true}, tab = {paper}, } -
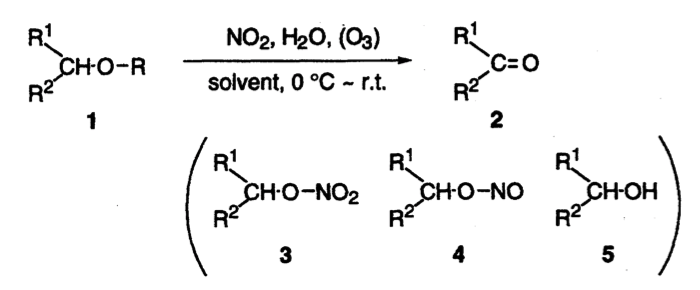 Direct Oxidation of Methyl Ethers to Carbonyl Compounds with a Combination of Nitrogen Dioxide and Water in the Presence or Absence of Ozone.Hitomi Suzuki*, Toyomi Takeuchi, and Tadashi MoriBull. Chem. Soc. Jpn., 1997, 70, 3111–3115.
Direct Oxidation of Methyl Ethers to Carbonyl Compounds with a Combination of Nitrogen Dioxide and Water in the Presence or Absence of Ozone.Hitomi Suzuki*, Toyomi Takeuchi, and Tadashi MoriBull. Chem. Soc. Jpn., 1997, 70, 3111–3115.A combination of nitrogen dioxide and water has been found to provide a new agent for the transformation of various alkyl methyl ethers 1 (R = Me) to carbonyl compounds 2 under mild conditions. The oxidation can be achieved successfully in dichloromethane, but hexafluoro-2-propanol was found to be most satisfactory as the solvent. The reaction was generally clean, and simple evaporation gave the expected oxidation product 2 in moderate to good yield. In the presence of ozone, but without water, the similar oxidation was observed only after prolonged reaction time, suggesting that some initial minor reaction with a strong oxidant, possible nitrogen trioxide, afforded an acidic promoter, which then worked as a catalyst similar to that involved in the reaction with a nitrogen dioxide and water system.
@article{suzuki1997direct, title = {Direct Oxidation of Methyl Ethers to Carbonyl Compounds with a Combination of Nitrogen Dioxide and Water in the Presence or Absence of Ozone.}, author = {Suzuki, Hitomi and Takeuchi, Toyomi and Mori, Tadashi}, journal = {Bull. Chem. Soc. Jpn.}, volume = {70}, number = {12}, pages = {3111--3115}, year = {1997}, publisher = {CSJ}, doi = {10.1246/bcsj.70.3111}, url = {https://doi.org/10.1246/bcsj.70.3111}, dimensions = {true}, tab = {paper}, } -
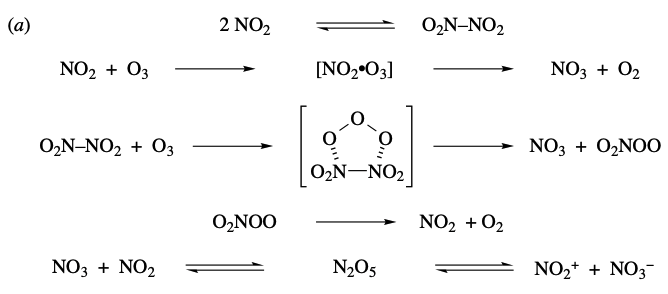 Ozone-mediated nitration of bicumene and derivatives with nitrogen dioxide. Preferential mesolytic bond cleavage over nuclear nitration in evidence for the electron transfer nature of the kyodai-nitration of arenesHitomi Suzuki* and Tadashi MoriJ. Chem. Soc., Perkin Trans. 2, 1997, 70, 1265–1274.
Ozone-mediated nitration of bicumene and derivatives with nitrogen dioxide. Preferential mesolytic bond cleavage over nuclear nitration in evidence for the electron transfer nature of the kyodai-nitration of arenesHitomi Suzuki* and Tadashi MoriJ. Chem. Soc., Perkin Trans. 2, 1997, 70, 1265–1274.The ozone-mediated reaction of bicumene and some derivatives 1 with nitrogen dioxide in dichloromethane at low temperatures resulted in the facile cleavage of the central C–C bond to give the corresponding benzyl nitrate and its descendants 4–6. Mesolytic bond cleavage occurred almost exclusively over nuclear substitution at temperatures as low as -20 °C, especially at low concentration (2 × 10-3 mol dm-3). This result may be rationalized in terms of initial electron transfer between the aromatic substrate and nitrogen trioxide generated in situ to form the aromatic radical cation, which then undergoes C–C bond scission at the benzylic position. In contrast, bibenzyl 2a is simply nitrated on the aromatic ring under similar conditions, giving the expected nitration products 7 and 8a–c along with a small amount of benzaldehyde 9. Results obtained from semi-empirical calculations and cyclic voltammetry are also in accord with the electron transfer nature of the reaction. The C–Si bond fission of benzyltrimethylsilane, C–C bond fragmentation of cyclic acetals of aromatic carbonyl compounds as well as side-chain reactions of toluene and derivatives, have all previously been observed under certain conditions of the kyodai-nitration and can be understood on a similar basis as described above. The possible involvment of electron transfer processes in aromatic nitration with acetyl nitrate has also been suggested.
@article{suzuki1997ozone, title = {Ozone-mediated nitration of bicumene and derivatives with nitrogen dioxide. Preferential mesolytic bond cleavage over nuclear nitration in evidence for the electron transfer nature of the kyodai-nitration of arenes}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 2}, number = {7}, pages = {1265--1274}, year = {1997}, publisher = {Royal Society of Chemistry}, doi = {10.1039/a700326i}, url = {https://doi.org/10.1039/a700326i}, dimensions = {true}, tab = {paper}, }
1996
-
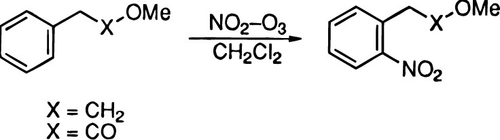 Ozone-mediated nitration of phenylalkyl ethers, phenylacetic esters, and related compounds with nitrogen dioxide. The highest ortho substitution observed in the electrophilic nitration of arenesHitomi Suzuki*, Toyomi Takeuchi, and Tadashi MoriJ. Org. Chem., 1996, 61, 5944–5947.
Ozone-mediated nitration of phenylalkyl ethers, phenylacetic esters, and related compounds with nitrogen dioxide. The highest ortho substitution observed in the electrophilic nitration of arenesHitomi Suzuki*, Toyomi Takeuchi, and Tadashi MoriJ. Org. Chem., 1996, 61, 5944–5947.By the combined action of ozonized oxygen and nitrogen dioxide (the kyodai-nitration), the title compounds were smoothly nitrated in dichloromethane at subzero degrees with high ortho positional selectivity. Although the conventional nitration of phenylacetic acid and esters mainly produces m- and p-nitro derivatives, the present nitration offers a simple high-yield synthesis of o-nitro derivatives which are important as precursor in organic synthesis. The proportions of the ortho isomer in the nitration products from methyl 2-phenylethyl ether and methyl phenylacetate were 71 and 88%, respectively, the latter value being the highest ortho isomer proportion so far observed in the electrophilic aromatic nitration. The observed high ortho selectivity has been rationalized in terms of radical cation intermediate and six-membered cyclic transition state.
@article{suzuki1996ozone, title = {Ozone-mediated nitration of phenylalkyl ethers, phenylacetic esters, and related compounds with nitrogen dioxide. The highest ortho substitution observed in the electrophilic nitration of arenes}, author = {Suzuki, Hitomi and Takeuchi, Toyomi and Mori, Tadashi}, journal = {J. Org. Chem.}, volume = {61}, number = {17}, pages = {5944--5947}, year = {1996}, publisher = {ACS Publications}, doi = {10.1021/jo9606867}, url = {https://doi.org/10.1021/jo9606867}, dimensions = {true}, tab = {paper}, } -
 Preference of the mesolytic cleavage over the nuclear substitution observed in the ozone-mediated nitration of bicumene with nitrogen dioxide. Implication to the electron transfer nature of the Kyodai-nitration of arenesHitomi Suzuki* and Tadashi MoriChem. Lett., 1996, 25, 647–648.
Preference of the mesolytic cleavage over the nuclear substitution observed in the ozone-mediated nitration of bicumene with nitrogen dioxide. Implication to the electron transfer nature of the Kyodai-nitration of arenesHitomi Suzuki* and Tadashi MoriChem. Lett., 1996, 25, 647–648.Ozone-mediated reaction of bicumene with nitrogen dioxide in dichloromethane at low temperature results in almost complete mesolytic cleavage of the hydrocarbon, accompanied by little or no nuclear nitration, in accordance with operation of the electron transfer process involving the nitrogen trioxide as initial electrophile.
@article{suzuki1996preference, title = {Preference of the mesolytic cleavage over the nuclear substitution observed in the ozone-mediated nitration of bicumene with nitrogen dioxide. Implication to the electron transfer nature of the Kyodai-nitration of arenes}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {Chem. Lett.}, volume = {25}, number = {8}, pages = {647--648}, year = {1996}, publisher = {Oxford University Press}, doi = {10.1246/cl.1996.647}, url = {https://doi.org/10.1246/cl.1996.647}, dimensions = {true}, tab = {paper}, } -
 Iron (III)-catalysed nitration of non-activated and moderately activated arenes with nitrogen dioxide–molecular oxygen under neutral conditionsHitomi Suzuki*, Shuji Yonezawa, Nobuaki Nonoyama, and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1996, 25, 2385–2389.
Iron (III)-catalysed nitration of non-activated and moderately activated arenes with nitrogen dioxide–molecular oxygen under neutral conditionsHitomi Suzuki*, Shuji Yonezawa, Nobuaki Nonoyama, and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1996, 25, 2385–2389.In the presence of molecular oxygen and a catalytic amount of tris(pentane-2,4-dionato)iron(III), non-activated and moderately activated arenes, which include alkylbenzenes, halogenobenzenes, phenolic ethers, naphthalene and derivatives, can be nitrated with nitrogen dioxide at ice-bath temperature or below to give the corresponding nitro derivatives in fair to good yields. An electron-transfer mechanism has been proposed, where an activated NO2-FeIII complex plays a key role in the cyclic process for converting arenes into nitroarenes.
@article{suzuki1996iron, title = {Iron (III)-catalysed nitration of non-activated and moderately activated arenes with nitrogen dioxide--molecular oxygen under neutral conditions}, author = {Suzuki, Hitomi and Yonezawa, Shuji and Nonoyama, Nobuaki and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 1}, number = {19}, pages = {2385--2389}, year = {1996}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P19960002385}, url = {https://doi.org/10.1039/P19960002385}, dimensions = {true}, tab = {paper}, } -
 Ozone-mediated nitration of naphthalene and some methyl derivatives with nitrogen dioxide. Remarkable enhancement of the 1-nitro/2-nitro isomer ratio and mechanistic implicationsHitomi Suzuki and Tadashi MoriJ. Chem. Soc., Perkin Trans. 2, 1996, 25, 677–683.
Ozone-mediated nitration of naphthalene and some methyl derivatives with nitrogen dioxide. Remarkable enhancement of the 1-nitro/2-nitro isomer ratio and mechanistic implicationsHitomi Suzuki and Tadashi MoriJ. Chem. Soc., Perkin Trans. 2, 1996, 25, 677–683.Naphthalene, 1- and 2-methylnaphthalenes were smoothly nitrated with nitrogen dioxide at low temperatures in the presence of ozone to afford the corresponding vitro derivatives in high yields. For naphthalene, the 1-nitro/2-nitro isomer ratios were remarkably high, mostly ranging from 35 to 70. 1,4-Dimethylnaphthalene suffered extensive side-chain substitution under similar conditions. The enhanced regioselectivity as compared with conventional nitration has been interpreted in terms of the electron transfer mechanism involving the nitrogen trioxide as the initial electrophile.
@article{suzuki1996ozone2, title = {Ozone-mediated nitration of naphthalene and some methyl derivatives with nitrogen dioxide. Remarkable enhancement of the 1-nitro/2-nitro isomer ratio and mechanistic implications}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 2}, number = {4}, pages = {677--683}, year = {1996}, publisher = {Royal Society of Chemistry}, doi = {10.1039/P29960000677}, url = {https://doi.org/10.1039/P29960000677}, dimensions = {true}, tab = {paper}, } -
 Solid-Phase Nitration of Salts of Aromatic Carboxylic Acids Using Nitrogen Dioxide in the Presence of Ozone. Controlling the Reaction Rate and the Isomer Ratio Using Metal IonsHitomi Suzuki*, Nobuaki Nonoyama, J Tomaru, and Tadashi MoriRuss. J. Org. Chem., 1996, 32, 248–251.
Solid-Phase Nitration of Salts of Aromatic Carboxylic Acids Using Nitrogen Dioxide in the Presence of Ozone. Controlling the Reaction Rate and the Isomer Ratio Using Metal IonsHitomi Suzuki*, Nobuaki Nonoyama, J Tomaru, and Tadashi MoriRuss. J. Org. Chem., 1996, 32, 248–251.@article{suzuki1996solid, title = {Solid-Phase Nitration of Salts of Aromatic Carboxylic Acids Using Nitrogen Dioxide in the Presence of Ozone. Controlling the Reaction Rate and the Isomer Ratio Using Metal Ions}, author = {Suzuki, Hitomi and Nonoyama, Nobuaki and Tomaru, J and Mori, Tadashi}, journal = {Russ. J. Org. Chem.}, volume = {32}, issue = {2}, pages = {248--251}, year = {1996}, publisher = {Kluwer Academic Plenum Publisher}, doi = {10.1002/chin.199644066}, url = {https://doi.org/10.1002/chin.199644066}, dimensions = {true}, tab = {paper}, }
1995
-
 Unusual isomer distribution of dinitrobenzenes and nitrophenols formed as side products during the ozone-mediated nitration of benzene with nitrogen dioxide. Further evidence for the alternative mechanism of electrophilic nitration of arenesHitomi Suzuki* and Tadashi MoriJ. Chem. Soc. Perkin Trans. 2, 1995, 32, 41–44.
Unusual isomer distribution of dinitrobenzenes and nitrophenols formed as side products during the ozone-mediated nitration of benzene with nitrogen dioxide. Further evidence for the alternative mechanism of electrophilic nitration of arenesHitomi Suzuki* and Tadashi MoriJ. Chem. Soc. Perkin Trans. 2, 1995, 32, 41–44.Dinitrobenzenes and nitrophenols formed as side products in the title reaction leading to nitrobenzene show an isomer distribution that is significantly different from those observed in the conventional nitration using nitric acid or nitric acid–sulfuric acid, suggesting the operation of a non-classical nitration mechanism involving nitrogen trioxide as the initial electrophile.
@article{suzuki1995unusual, title = {Unusual isomer distribution of dinitrobenzenes and nitrophenols formed as side products during the ozone-mediated nitration of benzene with nitrogen dioxide. Further evidence for the alternative mechanism of electrophilic nitration of arenes}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {J. Chem. Soc. Perkin Trans. 2}, number = {1}, pages = {41--44}, year = {1995}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P29950000041}, url = {https://doi.org/10.1039/P29950000041}, dimensions = {true}, tab = {paper}, } -
 Aromatic Nitration under Neutral Conditions Using Nitrogen Dioxide and Ozonne as the Nitrating Agent. Application to Aromatic Acetals and Acylal.Hitomi Suzuki*, Shuji Yonezawa, and Tadashi MoriBull. Chem. Soc. J., 1995, 68, 1535–1544.
Aromatic Nitration under Neutral Conditions Using Nitrogen Dioxide and Ozonne as the Nitrating Agent. Application to Aromatic Acetals and Acylal.Hitomi Suzuki*, Shuji Yonezawa, and Tadashi MoriBull. Chem. Soc. J., 1995, 68, 1535–1544.Cyclic acetals derived from aromatic carbonyl compounds can be nitrated smoothly with nitrogen dioxide in ice-cooled dichloromethane or acetonitrile in the presence of ozone and magnesium oxide to give ortho- and para-nitro derivatives as the major product in good combined yields, the acetal ring as a protective group remaining almost intact. An acylal derived from benzaldehyde similarly undergoes nitration on the aromatic ring to give an isomeric mixture of three nitro compounds, in which the ortho and meta isomers predominate, while aromatic orthoesters are rapidly decomposed to give simply the parent esters. Ring nitration under neutral conditions has been interpreted in terms of a nonclassical mechanism, in which nitrogen trioxide is involved as the initial electrophile.
@article{suzuki1995aromatic, title = {Aromatic Nitration under Neutral Conditions Using Nitrogen Dioxide and Ozonne as the Nitrating Agent. Application to Aromatic Acetals and Acylal.}, author = {Suzuki, Hitomi and Yonezawa, Shuji and Mori, Tadashi}, journal = {Bull. Chem. Soc. J.}, volume = {68}, number = {6}, pages = {1535--1544}, year = {1995}, publisher = {CSJ}, doi = {10.1246/bcsj.68.1535}, url = {https://doi.org/10.1246/bcsj.68.1535}, dimensions = {true}, tab = {paper}, } -
 Ozone-mediated Nitration of Aromatic Compounds with Lower Oxides of Nitrogen (The Kyodai-Nitration)Tadashi Mori and Hitomi Suzuki*Synlett, 1995, 68, 383–392.
Ozone-mediated Nitration of Aromatic Compounds with Lower Oxides of Nitrogen (The Kyodai-Nitration)Tadashi Mori and Hitomi Suzuki*Synlett, 1995, 68, 383–392.A novel non-acid methodology for the preparative nitration of aromatic compounds with the lower oxides of nitrogen using a combination of ozonized oxygen or air and some third substance as the promoter is described. The reaction, referred to as the kyodai-nitration, is now subject to industrially based research as the promising pollution-free and energy-saving substitute for the century-old, yet currently ongoing commercial process based on the use of nitric acid-sulfuric acid.
@article{mori1995ozone, title = {Ozone-mediated Nitration of Aromatic Compounds with Lower Oxides of Nitrogen (The Kyodai-Nitration)}, author = {Mori, Tadashi and Suzuki, Hitomi}, journal = {Synlett}, number = {5}, pages = {383--392}, year = {1995}, publisher = {Thieme}, doi = {10.1055/s-1995-4979}, url = {https://doi.org/10.1055/s-1995-4979}, dimensions = {true}, tab = {review}, } -
 Ozone-mediated reaction of anilides and phenyl esters with nitrogen dioxide: enhanced ortho-reactivity and mechanistic implicationsHitomi Suzuki*, Atsuo Tatsumi, Taro Ishibashi, and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1995, 68, 339–343.
Ozone-mediated reaction of anilides and phenyl esters with nitrogen dioxide: enhanced ortho-reactivity and mechanistic implicationsHitomi Suzuki*, Atsuo Tatsumi, Taro Ishibashi, and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1995, 68, 339–343.In the presence of ozone, anilides 1 can be nitrated rapidly with nitrogen dioxide in chloroform at 0 °C to give a high proportion of ortho-nitro derivatives (ortho : para= 1.2–4.4) in good yields. The phenyl esters 15 can be similarly nitrated on the aromatic ring without significant cleavage of the ester bond, giving a mixture of isomeric nitro derivatives in which the ortho-isomer predominates (ortho : para= 1.1–1.5). The origin of the enhanced ortho reactivity is discussed in terms of an electron-transfer process involving the nitrogen trioxide as initial electrophile.
@article{suzuki1995ozone, title = {Ozone-mediated reaction of anilides and phenyl esters with nitrogen dioxide: enhanced ortho-reactivity and mechanistic implications}, author = {Suzuki, Hitomi and Tatsumi, Atsuo and Ishibashi, Taro and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 1}, number = {4}, pages = {339--343}, year = {1995}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P19950000339}, url = {https://doi.org/10.1039/P19950000339}, dimensions = {true}, tab = {paper}, } -
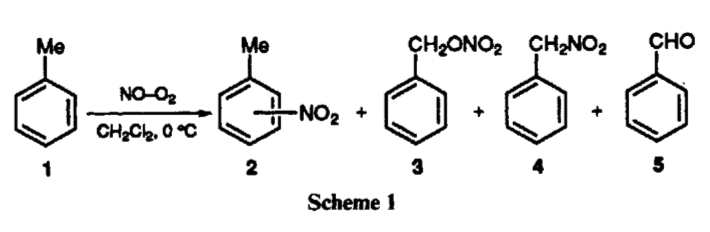 Nitration of nonactivated arenes with a ternary system NO–NO2–O2. Mechanistic implications of the Kyodai-nitrationHitomi Suzuki* and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1995, 68, 291–293.
Nitration of nonactivated arenes with a ternary system NO–NO2–O2. Mechanistic implications of the Kyodai-nitrationHitomi Suzuki* and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1995, 68, 291–293.The ternary mixture NO–NO2–O2 has been found to be more effective than the binary mixture NO2–O2 as a nitrating agent for nonactivated arenes, such as toluene and chlorobenzene; the results may be rationalized in terms of an initial electron transfer mechanism in which nitrogen trioxide, formed from nitrogen monoxide and dioxygen, oxidizes the arene to its radical cation.
@article{suzuki1995nitration, title = {Nitration of nonactivated arenes with a ternary system NO--NO2--O2. Mechanistic implications of the Kyodai-nitration}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 1}, number = {4}, pages = {291--293}, year = {1995}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P19950000291}, url = {https://doi.org/10.1039/P19950000291}, dimensions = {true}, tab = {paper}, }
1994
-
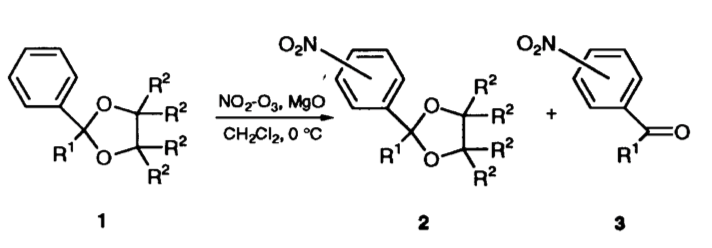 Ozone-mediated reaction of aromatic acetals and acylal with nitrogen dioxide: a novel methodology for the nuclear nitration of acid-sensitive aromatic compounds under neutral conditionsHitomi Suzuki*, Shuji Yonezawa, Tadashi Mori, and Koichi MaedaJ. Chem. Soc., Perkin Trans. 1, 1994, 68, 1367–1369.
Ozone-mediated reaction of aromatic acetals and acylal with nitrogen dioxide: a novel methodology for the nuclear nitration of acid-sensitive aromatic compounds under neutral conditionsHitomi Suzuki*, Shuji Yonezawa, Tadashi Mori, and Koichi MaedaJ. Chem. Soc., Perkin Trans. 1, 1994, 68, 1367–1369.Cyclic acetals 1c–e and 1g can be nitrated smoothly with nitrogen dioxide in ice-cooled dichloromethane in the presence of ozone and magnesium oxide to give mainly ortho- and para- nitro derivatives in good combined yields, the acetal protective group remaining almost intact; acylal 6 is similarly nitrated on the aromatic ring to give a mixture of isomeric nitro derivatives 7, in which the ortho and meta isomers predominate.
@article{suzuki1994ozone, title = {Ozone-mediated reaction of aromatic acetals and acylal with nitrogen dioxide: a novel methodology for the nuclear nitration of acid-sensitive aromatic compounds under neutral conditions}, author = {Suzuki, Hitomi and Yonezawa, Shuji and Mori, Tadashi and Maeda, Koichi}, journal = {J. Chem. Soc., Perkin Trans. 1}, number = {11}, pages = {1367--1369}, year = {1994}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P19940001367}, url = {https://doi.org/10.1039/P19940001367}, dimensions = {true}, tab = {paper}, } -
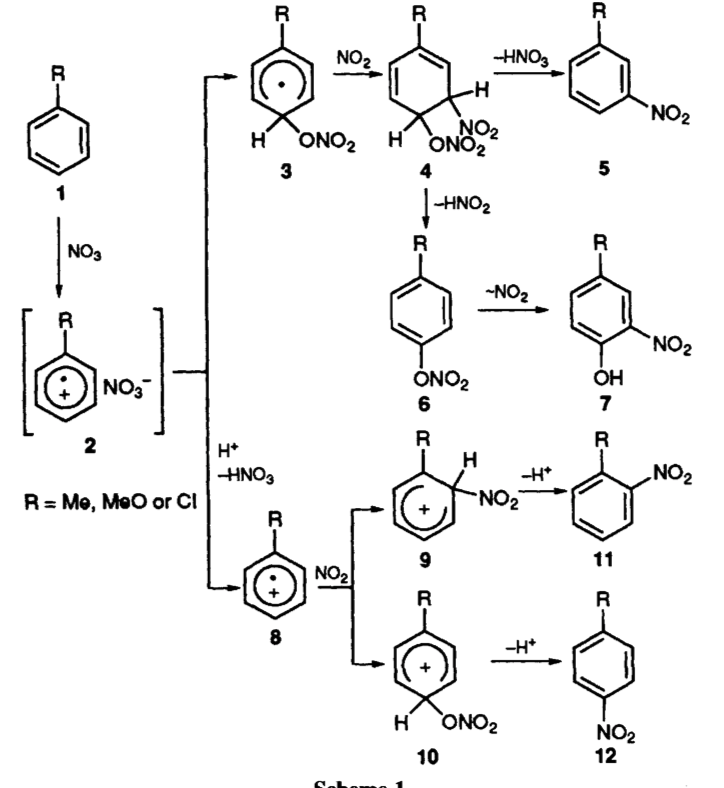 Ozone-mediated nitratrion of arenes with nitrogen dioxide: change-over of the orienting influences of alkyl, alkoxyl and halogen substituent groups from meta to ortho-para dominanceHitomi Suzuki*, Takashi Murashima, and Tadashi MoriJ. Chem. Soc., Chem. Commun., 1994, 68, 1443–1444.
Ozone-mediated nitratrion of arenes with nitrogen dioxide: change-over of the orienting influences of alkyl, alkoxyl and halogen substituent groups from meta to ortho-para dominanceHitomi Suzuki*, Takashi Murashima, and Tadashi MoriJ. Chem. Soc., Chem. Commun., 1994, 68, 1443–1444.In ozone-mediated nitration of toluene, anisole and chlorobenzene with nitrogen dioxide, the initial products have been found to be composed mainly of the meta-nitro derivatives, but the isomer distributions are rapidly replaced by the ortho-para isomers as the reaction proceeds, sugesting the operation of the electron transfer mechanism involving the nitrogen trioxide as initial electrophile.
@article{suzuki1994ozone2, title = {Ozone-mediated nitratrion of arenes with nitrogen dioxide: change-over of the orienting influences of alkyl, alkoxyl and halogen substituent groups from meta to ortho-para dominance}, author = {Suzuki, Hitomi and Murashima, Takashi and Mori, Tadashi}, journal = {J. Chem. Soc., Chem. Commun.}, number = {12}, pages = {1443--1444}, year = {1994}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/C39940001443}, url = {https://doi.org/10.1039/C39940001443}, dimensions = {true}, tab = {paper}, } -
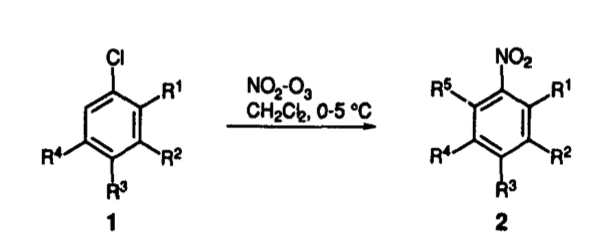 Ozone-mediated reaction of polychlorobenzenes and some related halogeno compounds with nitrogen dioxide: a novel non-acid methodology for the selective mononitration of moderately deactivated aromatic systemsHitomi Suzuki*, Tadashi Mori, and Koichi MaedaSynthesis, 1994, 1994, 841–845.
Ozone-mediated reaction of polychlorobenzenes and some related halogeno compounds with nitrogen dioxide: a novel non-acid methodology for the selective mononitration of moderately deactivated aromatic systemsHitomi Suzuki*, Tadashi Mori, and Koichi MaedaSynthesis, 1994, 1994, 841–845.In the presence of ozone and preferably methanesulfonic acid as catalyst, polychlorobenzenes undergo selevtive mononitration with nitrogen dioxide at low temperatures, giving the corresponding polychloronitrobenzenes, in most cases in nearly quantitative yields.
@article{suzuki1994ozone3, title = {Ozone-mediated reaction of polychlorobenzenes and some related halogeno compounds with nitrogen dioxide: a novel non-acid methodology for the selective mononitration of moderately deactivated aromatic systems}, author = {Suzuki, Hitomi and Mori, Tadashi and Maeda, Koichi}, journal = {Synthesis}, volume = {1994}, number = {08}, pages = {841--845}, year = {1994}, publisher = {Georg Thieme Verlag}, doi = {10.1055/s-1994-25586}, url = {https://doi.org/10.1055/s-1994-25586}, dimensions = {true}, tab = {paper}, } -
 Ozone-mediated nitration of chloro-and bromo-benzenes and some methyl derivatives with nitrogen dioxide. High ortho-directing trends of the chlorine and bromine substituentsHitomi Suzuki* and Tadashi MoriJ. Chem. Soc., Perkin Trans. 2, 1994, 1994, 479–484.
Ozone-mediated nitration of chloro-and bromo-benzenes and some methyl derivatives with nitrogen dioxide. High ortho-directing trends of the chlorine and bromine substituentsHitomi Suzuki* and Tadashi MoriJ. Chem. Soc., Perkin Trans. 2, 1994, 1994, 479–484.In the presence of ozone, chloro- and bromo-benzenes are nitrated smoothly with nitrogen dioxide at low temperatures, giving a mixture of the corresponding nitro derivatives in nearly quantitative yield. The nitration products are generally ortho-rich as compared with those obtained by the conventional procedures based on the use of nitric acid or mixed acid. Interestingly, the ortho : para isomer ratios of the products can be reversed from ortho-rich (o : p= 1.14 and 1.09) to para-predominant (o : p= 0.45 and 0.68) simply by altering the initial concentration of the substrate. This enigmatic phenomenon has been interpreted in terms of the equilibrium between the monomer and the dimer forms of the cation radical derived from a halogenobenzene and the difference in their relative reactivity toward nitrogen dioxide. Similar preference for substitution ortho to the halogen substituent has been observed with 4-chloro- and 4-bromo-toluenes, which concurrently suffer extensive ipso attack by the present reagent system, leading to the formation of 4-methyl-2-nitrophenol as a common side product.
@article{suzuki1994ozone4, title = {Ozone-mediated nitration of chloro-and bromo-benzenes and some methyl derivatives with nitrogen dioxide. High ortho-directing trends of the chlorine and bromine substituents}, author = {Suzuki, Hitomi and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 2}, number = {3}, pages = {479--484}, year = {1994}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P29940000479}, url = {https://doi.org/10.1039/P29940000479}, dimensions = {true}, tab = {paper}, }
1993
-
 Ozone-mediated nitration of alkylbenzenes and related compounds with nitrogen dioxideHitomi Suzuki, Takashi Murashima, Iku Kozai, and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1993, 1994, 1591–1597.
Ozone-mediated nitration of alkylbenzenes and related compounds with nitrogen dioxideHitomi Suzuki, Takashi Murashima, Iku Kozai, and Tadashi MoriJ. Chem. Soc., Perkin Trans. 1, 1993, 1994, 1591–1597.In the presence of ozone, nitrogen dioxide exhibits a strong nitrating ability for alkylbenzenes at low temperatures, converting them into the corresponding nitro derivatives in high yield. The addition of a protonic acid as catalyst enhances considerably the ability of this nitrating system and leads to a good yield of polynitro compounds. The reaction is clean and proceeds rapidly without any accompanying side-chain substitution or aryl–aryl coupling. It shows no kinetic dependence on the concentration of substrates and, as far as can be judged from relative reactivities and isomer distributions of products, it gives the appearance of being an electrophilic aromatic process. A possible role for nitrogen trioxide has been suggested as the initial electrophilic agent for the nitration of alkylbenzenes.
@article{suzuki1993ozone, title = {Ozone-mediated nitration of alkylbenzenes and related compounds with nitrogen dioxide}, author = {Suzuki, Hitomi and Murashima, Takashi and Kozai, Iku and Mori, Tadashi}, journal = {J. Chem. Soc., Perkin Trans. 1}, number = {14}, pages = {1591--1597}, year = {1993}, publisher = {The Royal Society of Chemistry}, doi = {10.1039/P19930001591}, url = {https://doi.org/10.1039/P19930001591}, dimensions = {true}, tab = {paper}, }